- (12.10) Производитель должен документально оформить общую политику в отношении валидации, ее задачи и принципы, включая валидацию технологических процессов, процедур очистки, аналитических методик, процедур контроля в процессе производства, компьютеризированных систем и в отношении лиц, ответственных за разработку, проверку, утверждение и документальное оформление каждого этапа валидации.
- (12.11) Критические параметры и (или) характеристики, как правило, следует определять на стадии разработки или на основании данных предварительного опыта работы; следует также определить диапазоны значений этих критических параметров и (или) характеристик, необходимые для обеспечения воспроизводимости процесса. При этом необходимо:
- определить критические характеристики ФС как продукции;
- указать параметры процесса, которые могут влиять на критические показатели качества ФС;
- установить диапазон значений каждого критического параметра процесса, который предполагается использовать при серийном производстве и контроле процесса.
- (12.12) Операции, которые считаются критическими для качества и чистоты ФС, подлежат валидации.
Документация по валидации (12.2)
- (12.20) Для каждого процесса, подлежащего валидации, должен быть разработан протокол валидации. Этот протокол должен быть проверен и утвержден подразделением (подразделениями) качества и другими соответствующими подразделениями.
- (12.21) В протоколе валидации должны быть определены критические стадии процесса и критерии приемлемости, а также вид проводимой валидации (например, ретроспективная, перспективная, сопутствующая) и количество производственных циклов.
- (12.22) Отчет о валидации должен содержать перекрестные ссылки на протокол валидации и обобщать полученные результаты, объяснять любые обнаруженные отклонения с соответствующими выводами, включающими рекомендуемые изменения для исправления недостатков.
- (12.23) Любые отклонения от протокола валидации должны быть оформлены документально с соответствующим обоснованием.
- (12.30) До начала работ по валидации процесса необходимо завершить квалификацию критического оборудования и вспомогательных систем. Квалификацию обычно проводят по следующим этапам (по отдельности или в совокупности):
- квалификация проекта: документально оформленное подтверждение того, что предложенный проект производственных помещений, оборудования или систем является пригодным для применения по назначению.
- квалификация монтажа: документально оформленное подтверждение того, что монтаж помещений, систем и оборудования (установленных или модифицированных) выполнен в соответствии с утвержденным проектом, рекомендациями изготовителя и (или) требованиями производителя лекарственных средств.
- квалификация функционирования: документально оформленное подтверждение того, что помещения, системы и оборудование (установленные или модифицированные) функционируют в соответствии со своим предназначением во всех предусмотренных режимах работы.
- квалификация эксплуатации: документально оформленное подтверждение того, что помещения, системы и оборудование при совместном использовании работают эффективно и с воспроизводимыми показателями в соответствии с установленными требованиями и характеристиками процесса.
Подходы к валидации процесса (12.4)
- а) (1) определены критические показатели качества и критические параметры процесса;
- б) (2) установлены надлежащие критерии приемлемости и контроля в процессе производства;
- в) (3) отсутствовали существенные сбои в ходе процесса или брак продукции по причинам, не связанным с ошибками оператора или отказами оборудования;
- г) (4) были установлены профили примесей для данной ФС.
- (12.45) Серии, отобранные для ретроспективной валидации, должны представлять репрезентативную выборку из всех серий, произведенных за проверяемый период, в том числе любых серий, не соответствующих спецификациям. При этом количество таких серий должно быть достаточным для доказательства постоянства процесса. В целях получения данных для ретроспективной валидации процесса может быть проведено испытание архивных образцов.
Программа валидации процесса (12.5)
- (12.50) Количество производственных циклов, необходимых для валидации, должно зависеть от сложности процесса или от значимости изменений процесса, подлежащих рассмотрению. Для перспективной и сопутствующей валидации должны быть использованы данные, полученные для трех последовательных производственных серий продукции надлежащего качества. Однако могут быть ситуации, когда для доказательства постоянства процесса необходимы дополнительные производственные циклы (например, процессы производства сложных ФС или длительные процессы производства ФС). Для оценки постоянства процесса при ретроспективной валидации, как правило, необходимо исследовать данные для 10 — 30 последовательных серий, но при соответствующем обосновании это число может быть уменьшено.
- (12.51) Во время проведения исследований по валидации процесса необходимо контролировать и проверять его критические параметры. Параметры процесса, не связанные с качеством, например, переменные, контролируемые в целях сокращения потребления энергии или использования оборудования, можно не включать в валидацию процесса.
- (12.52) Валидация процесса должна подтверждать, что профиль примесей для каждой ФС находится в заданных пределах. Профиль примесей должен быть сходен (либо быть лучше) с ранее полученным профилем, а также (где это применимо) с профилем примесей, установленным при разработке процесса или серий, использованных для основных клинических и токсикологических исследований.
Периодическая проверка валидированных систем (12.6)
- (12.60) Системы и процессы необходимо подвергать периодической оценке для подтверждения того, что они по-прежнему функционируют правильным образом. Если в процесс или систему не было внесено существенных изменений и обзор качества подтвердил, что система или процесс постоянно обеспечивают производство материала, соответствующего спецификациям, как правило, отсутствует необходимость в проведении повторной валидации.
- (12.70) Процедуры очистки, как правило, должны пройти валидацию. Валидацию очистки проводят в случаях, при которых контаминация или перенос веществ представляют наибольшую опасность для качества ФС. Например, на начальных стадиях технологического процесса может не требоваться проведения валидации процедур очистки оборудования, если остаточные вещества удаляют на последующих стадиях очистки.
- (12.71) Валидация процедур очистки должна отражать фактический характер использования оборудования. Если разные ФС или различную промежуточную продукцию производят на одном и том же оборудовании и это оборудование очищают одним и тем же способом, то для валидации очистки можно выбрать репрезентативную промежуточную продукцию или ФС. Такой выбор должен основываться на данных о растворимости и трудностях очистки, а также на расчете предельного содержания остатков, с учетом их активности, токсичности и стабильности.
- (12.72) В протоколе валидации очистки должны быть описаны оборудование, подлежащее очистке, процедуры, материалы, приемлемые уровни очистки, контролируемые и регулируемые параметры и аналитические методики. В протоколе необходимо также указать виды отбираемых проб, способы их отбора и маркировки.
- (12.73) Для обнаружения как нерастворимых, так и растворимых остатков методы отбора проб должны включать, исходя из ситуации, взятие мазков, смывов или другие методы (например, прямую экстракцию). Используемые методы отбора проб должны позволять количественно определять уровни остатков на поверхностях оборудования после очистки. Метод отбора проб посредством взятия мазков может оказаться практически неосуществимым, если контактирующие с продуктом поверхности являются труднодоступными вследствие конструктивных особенностей оборудования (например, внутренние поверхности шлангов, транспортных трубопроводов, емкости реакторов с узкими люками, а также небольшое по размеру сложное оборудование, например, микронизаторы и микрораспылители) и (или) если существуют ограничения процесса (например, обработка токсичных веществ).
- (12.74) Необходимо использовать валидированные аналитические методики, обладающие достаточной чувствительностью для обнаружения остатков или контаминантов. Предел обнаружения каждой аналитической методики должен быть достаточным для обнаружения определенного приемлемого уровня остатка или контаминанта. Для методики необходимо установить достигаемый уровень извлечения вещества. Пределы содержания остатков должны быть реальными, достижимыми, проверяемыми и основываться на содержании наиболее вредного остатка. Пределы можно устанавливать, основываясь на минимальном обладающем известной фармакологической, токсикологической или физиологической активностью количестве ФС или ее наиболее вредного компонента.
- (12.75) Для процессов, в которых существует необходимость снижения общего количества микроорганизмов или эндотоксинов в ФС, или для других процессов, где может иметь значение такая контаминация (например, производство нестерильных ФС, используемых для производства стерильных лекарственных препаратов), исследование очистки и (или) санитарной обработки оборудования необходимо проводить в отношении контаминации микроорганизмами и эндотоксинами.
- (12.76) Производитель должен контролировать процедуры очистки с определенной периодичностью после валидации, чтобы убедиться, что эти процедуры являются эффективными при их использовании во время текущего технологического процесса. Чистоту оборудования, когда это осуществимо, необходимо контролировать посредством проведения аналитических испытаний и визуального осмотра. Визуальный осмотр позволяет обнаружить значительные скопления контаминантов на небольших участках, которые могут оказаться не обнаруженными иным способом при отборе проб и (или) анализе.
Валидация аналитических методик (12.8)
- (12.80) Используемые аналитические методики должны пройти валидацию. Пригодность всех используемых методик испытаний необходимо, тем не менее, проверять в реальных условиях применения, а результаты оформлять документально.
- (12.81) Валидация методик должна проводиться с учетом характеристик, приведенных в руководствах по валидации аналитических методик. Объем проводимой аналитической валидации должен зависеть от цели анализа и стадии технологического процесса производства ФС.
- (12.82) До начала валидации аналитических методик должна быть проведена соответствующая квалификация аналитического оборудования.
539. (12.83) Необходимо вести полные записи любых изменений валидированной аналитической методики. Такие записи должны отражать причину изменения и соответствующие данные для подтверждения того, что изменение приводит к результатам, которые столь же точны и надежны, как и результаты, полученные с помощью принятой методики.
Валидация процессов. Новый подход FDA.
Попов А.Ю. ООО «Эй Пи Интернэйшнл»
Введение
18 ноября 2008 г. произошло важное событие для всех, кто производит фармацевтическую продукцию. В США был опубликован проект нового руководства FDA по валидации процессов при производстве лекарственных средств (Guidance for Industry. Process Validation: General Principles and Practices) (1). Предыдущий аналогичный документ FDA был опубликован в мае 1987 г., т.е. более 30 лет тому назад. В новом руководстве нашли отражение современные тенденции, относящиеся к обеспечению качества лекарственных средств, и сформулированные в инициативе FDA, опубликованной в 2002 г., и получившей название: «Фармацевтические правила cGMP в 21 веке. Подход, основанный на анализе рисков.» (“Pharmaceutical cGMPs for the 21 st Century: Risk-Based Approach”) (2). В этой инициатива впервые был провозглашен новый системный подход к обеспечению качества лекарственных средств, основанный на анализе и управлении рисками (Risk Based Approach), а также на применении системы мониторинга технологического процесса с использованием новейших аналитических средств (Process Analytical Technology -PAT), и на создании всеобъемлющей системы качества (Quality System) производства лекарственных средств. Этот подход нашел отражение в серии новых руководств FDA, вышедших в свет в 2004 и 2006 годах. (3, 4, 5).
Проведение валидации процессов и квалификации оборудования с использованием анализа рисков рассматривалось в предыдущих публикациях журнала «Чистые помещения и технологические среды» (6, 7, 8). Настоящая статья призвана познакомить российских специалистов с содержанием нового документа FDA и с новыми тенденциями в области обеспечения качества фармацевтической продукции.
Область применения руководства FDA
Новое руководство FDA по валидации процессов применимо к производству следующих категорий лекарственных препаратов:
Лекарственные средства, предназначенные для людей;
Ветеринарные препараты;
Биологические и биотехнологические продукты;
Активные фармацевтические ингредиенты и фармацевтические субстанции;
Лекарственные средства, произведенные в комбинации с медицинскими изделиями.
Хотелось бы обратить внимание читателей на то, что новое руководство для промышленности (Guidance or Industry), как и все другие руководства, издаваемые FDA, носит не обязательный, а рекомендательный характер. Оно выражает современные представления и подходы к валидации процессов, которых придерживается FDA. Обязательными являются так называемые правила cGMP (current Good manufacturing Practice), которые имеют силу закона и публикуются в виде Code Federal Register (CFR).
Философия нового руководства FDA по валидации процессов.
Американские cGMP, которые носят обязательный характер и имеют силу закона, требуют, чтобы валидированное (validating) фармацевтическое производство осуществляло выпуск лекарственных средства с высоким уровнем обеспечения качества (21 CFR 211.100(a) и 211.110 (а)). При этом указывается, что валидация процессов относится к деятельности по обеспечению качества фармацевтической продукции. Главной задачей обеспечения качества является то, чтобы лекарственные средства были изготовлены годными для применения в соответствии с их назначением (intended use). Под годностью понимается безопасность и эффективность лекарственных препаратов при приеме пациентом.
Обеспечение качества, согласно cGMP, базируется на выполнении следующих основополагающих принципов:
- Качество, безопасность и эффективность заложены или «встроены» (built into) в лекарственный препарат на стадии его разработки;
Качество препарата не может быть гарантировано только за счет проверки качества полупродуктов и готовой продукции;
Каждый шаг производственного процесса лекарственных средств постоянно находится под контролем (управлением) для обеспечения того, чтобы готовая продукция полностью соответствовала всем показателям качества, указанным в спецификации на нее.
Базируясь на этих трех принципах, новое руководство FDA дает следующее определение валидации процессов:
Валидацией процесса называется сбор и анализ данных, которые начинаются на стадии разработки процесса и продолжаются на стадии промышленного производства для того, чтобы получить научно-обоснованное доказательство, что процесс способен стабильно производить качественную продукцию.
Валидация процесса включает в себя определенную последовательность действий, выполняемых на протяжении жизненного цикла фармацевтического продукта и процесса его производства. При этом понятие «жизненный цикл» (product lifecycle) продукта понимается так, как оно определено в Руководстве ICH Q8A Pharmaceutical Development (9).
Новое руководство FDA выделяет на три основных стадии валидации процесса:
Стадия 1. - Разработка процесса. Процесс промышленного (коммерческого) производства оценивается (валидируется) на этой стадии, основываясь на знании процесса, полученном при его разработке и масштабировании.
Стадия 2. - Квалификация процесса. На этой стадии производственный процесс проверяется на способность устойчиво производить качественную продукцию при промышленном (коммерческом) производстве.
Стадия 3. - Продолжающаяся верификация процесса: Она включает в себя регулярные проверки, выполняемые в процессе текущего производства, для подтверждения того, что процесс находится под контролем (т.е. обеспечивается гарантированное производство продукции надлежащего качества).
При этом подчеркивается, что перед тем, как любая партия лекарственного препарата будет отгружена потребителю, производителю следует получить высокую степень гарантии того, что производственный процесс будет устойчиво давать продукцию, соответствующую всем показателям качества. Этой гарантии следует добиваться на основании объективной информации, получаемой на стадиях лабораторной, пилотной и/или промышленной отработки процесса. С помощью этой информации следует продемонстрировать, что процесс промышленного (коммерческого) производства способен устойчиво (стабильно) производить готовую продукцию надлежащего качества в реальных условиях промышленного производства, включая такие условия как «наихудший случай» (“worst case”), при которых имеет место высокий риск нарушения требований производственного процесса.
Успешная валидация процесса зависит от наличия информации и знаний о природе процесса. Эти знания являются залогом того, что производственный процесс будет управляться надлежащим образом. Каждому производителю следует добиваться высокой степени понимания процесса для надлежащего обеспечения выпуска качественной продукции.
Каждый процесс выполняется в условиях наличия внешних и внутренних вариаций (переменных значений технологических параметров). Эти вариации должны быть в определенных пределах, чтобы гарантировать стабильный выпуск продукции надлежащего качества. Т.е. валидированный производственный процесс должен, не зависимо от этих вариаций, обеспечить выпуск качественных лекарственных средств. Для этого производителю следует:
Понимать причины и источники вариаций;
Выявлять наличие и степень вариаций;
Понимать влияние вариаций на процесс и, соответственно, на показатели качества продукта;
Управлять этими вариациями в зависимости от того, какое влияние они оказывают на процесс и продукт.
Эти знания получаются на стадии разработки процесса, уточняются и дополняются на стадиях масштабирования процесса и внедрения его в промышленное производство.
Главная задача на стадии разработки процесса и его внедрения в производство - получить поле допустимых значений всех технологических параметров и отработать меры контроля по удержанию процесса в этом поле.
После отработки и внедрения производственного процесса, производитель должен поддерживать процесс в поле допустимых значений технологических параметров на протяжении всего времени использования процесса.
Итак, валидация процесса является частью системы обеспечения качества готовой продукции. Ее цель дать уверенность всем заинтересованным сторонам (производителю, контролирующим органам и потребителю) в том, что производственный процесс надлежащим образом управляется (находится под контролем) и гарантирует выпуск продукции надлежащего качества, несмотря на возможные риски, которые связаны с сырьем и материалами, производственной средой, оборудованием и персоналом.
Современные требования GMP предполагают, что, создавая систему обеспечения качества, производителю следует документально подтвердить, что все риски, способные оказать негативное влияние на качество продукции, выявлены, проанализированы и разработана система предупредительных и корректирующих действий, позволяющая удерживать эти риски под контролем. В этом, собственно, и состоит «основанный на рисках подход» (risk based approach) к обеспечению качества лекарственных средств.
Валидации процесса должна собрать объективные доказательства для такого документального подтверждения. Следовательно, валидация процесса должна проводиться с учетом выявленных рисков, включая проведение валидационных испытаний в ситуации, относящейся к «наихудшему случаю» (worst case). При этом под «наихудшим случаем» понимается возможная ситуация, возникающая в производстве лекарственных средств, при которой технологические параметры, условия производства, функционирование оборудования и действия персонала образуют комбинацию эффектов, наихудшую для производства продукции надлежащего качества.
Кратко философию нового руководства можно сформулировать следующим образом.
- Валидация процесса начинается уже на стадии его разработки. Полностью процесс валидируется при внедрении его в коммерческое производство. Затем периодически проверяется его валидированность, т.е. способность устойчиво производить продукцию надлежащего качества.
- С самого начала разработки процесса необходимо проводить анализ рисков, способных помешать выпуску продукции надлежащего качества. Систему управления процессом (контроль процесса) следует создавать с учетом выявленных рисков. При этом валидация процесса также проводится с учетом выявленных рисков, в том числе и для ситуаций относящихся к «наихудшему случаю».
Новое руководство содержит целый ряд практических рекомендаций. Они касаются последовательных стадий валидации процесса, выполняемых на протяжении всего жизненного цикла фармацевтической продукции.
Согласно современному подходу к обеспечению качества лекарственных препаратов, каждый фармацевтический продукт проходит четыре последовательных стадии жизненного цикла (10):
- Разработка продукта и процесса его производства (Product and Process Design);
- Внедрение продукта и процесса в коммерческое производство (Technology Transfer);
- Коммерческое производство продукта (Manufacturing);
- Снятие продукта с производства (Product Descontinuation).
Стадия 1. Разработка фармацевтического продукта и процесса его производства
На первой стадии жизненного цикла производится сбор данных о процессе и достигается его глубокое понимание (Process Knowledge and Understanding). Определяется совокупность оптимальных значений технологических параметров, с помощью которых гарантируется получение продукта заданного качества. С помощью анализа рисков выявляются критические этапы производственного процесса и критические технологические параметры.
Также определяется стратегия управления производственным процессом (Process Control), которая позволит эффективно и надежно поддерживать технологические параметры в заданной области значений.
Новое руководство рекомендует на этой стадии использовать такие инструменты, как анализ рисков (например, с помощью системы НАССР), в также планирование эксперимента (Design of Experiments -DOE). С их помощью можно существенно сократить объем и продолжительность исследований, как в лабораторном, так и в полупромышленном (пилотном) масштабе. Более того, они позволяют эффективно выявить изменчивость и взаимозависимость технологических параметров, а также выработать эффективные меры контроля критических этапов процесса.
От разработчиков требуется предоставить многократно проверенные (статистически достоверные) доказательства того, что, несмотря на любые допустимые изменения сырья, материалов, технологических сред (вода, пар, сжатый воздух), качества производственной среды, технологических параметров, персонала и др., процесс будет гарантированно обеспечивать продукцию надлежащего качества. Для этого разработчику сначала требуется определить:
а) научно и экспериментально обоснованные количественные и качественные требования (диапазоны допустимых значений) ко всем элементам производственного процесса, соблюдение которых позволит гарантировать выпуск продукции надлежащего качества;
б) перечень контрольных мер (мер по управлению процессом), с помощью которых обеспечивается удержание всех элементов производственного процесса внутри их диапазонов допустимых значений, и, тем самым, гарантируется выпуск продукции надлежащего качества;
Именно эти две составляющих технологической разработки образуют, так называемое, технологическое пространство процесса (Design Space).
Технологическое пространство процесса содержит, с одной стороны, требования ко всем элементам технологического процесса, т.е. к сырью и материалам, помещениям, оборудованию и технологическим средам, производственной среде, всем этапам технологического процесса, персоналу и др., а с другой - меры управления процессом, направленные на гарантированное обеспечение надлежащего качества продукции.
Самым тщательным образом следует отрабатывать именно контрольные функции. Для этого необходимо проводить испытания в искусственно создаваемых ситуациях (наихудший случай- Worst Case), которые соответствуют наихудшим (граничным) значениям допустимых диапазонов технологических параметров. Эффективность мер контроля (управления) технологическим процессом следует многократно проверять для получения надежного результата.
Чтобы свести к минимуму число повторных испытаний, уже на ранних стадиях в исследованиях и разработках рекомендуется применять планирование экспериментов (Design of Experiments - DOE). DOE позволяет получить статистически достоверную информацию о взаимной зависимости различных технологических параметров и значительно сократить объем и продолжительность работ. DOE также дает возможность кротчайшим путем выявить область оптимальных значений технологических параметров.
Рекомендуется применять и различные инструменты анализа рисков, в частности, систему НАССР (Hazard Analysis and Critical Control Points). Достоинство этой системы состоит в том, что она представляет собой научный и системный подход к управлению производством, гарантированно обеспечивающий выпуск продукции надлежащего качества. Она сокращает объем работ за счет того, что работает с главным и игнорирует множество второстепенного. Ее применение обеспечивает новое видение и понимание технологического процесса. Система НАССР позволяет надежно и эффективно найти все критические этапы производственного процесса, выявить опасные факторы, способные помешать выпуску продукции надлежащего качества, определить критические (допустимые) пределы технологических параметров, создать систему мониторинга и разработать комплекс предупредительных и корректирующих действий (Corrective and Preventive Actions-CAPA), с помощью которых обеспечивается управление процессом (11, 12, 13, 14).
Весь процесс технологических исследований и разработок рекомендуется строго документировать в соответствии с надлежащей практикой документирования (Good Documentation Practice)(9).
Стадия 2. Внедрение процесса в производство
Вторая стадия жизненного цикла процесса является своеобразным мостиком между стадией разработок и стадией производства. Она решает две главные задачи:
Масштабирование процесса;
Проведение валидации процесса, которая в новом руководстве названа квалификацией (Process Qualification) и, сопутствующее ей, уточнение технологического пространства, устанавливаемого для коммерческого производства.
Цель квалификации процесса состоит в том, чтобы показать, что процесс способен обеспечить устойчивое коммерческое производство, т.е. готов гарантировать качество продукта, несмотря на любую изменчивость характеристик сырья и материалов, а также технологических показателей процесса. Эта работа проводится в границах, очерченных технологическим пространством, для подтверждения того, что это пространство определено правильно. Если это не подтверждается, то границы технологического пространства уточняются, и квалификация процесса повторяется.
Чтобы избежать потерь времени и средств на повторную квалификацию процесса, технологическое пространство процесса должно быть очень тщательно определено, а его границы многократно проверены, еще на стадии разработки процесса.
Работы по квалификации проекта состоят из двух составляющих:
Квалификация помещений, технологического оборудования и технологических сред;
Квалификация эксплуатации (PQ).
В новом руководстве подчеркивается, что выполнение этих работ должно обязательно предшествовать началу промышленного производства и реализации лекарственных средств.
Здания и сооружения. Надлежащее исполнение производственных зданий и сооружений подпадает под требования, содержащиеся в американских cGMP (21CFR part 211, subpart C “Buildings and Facilities”). Работы, проводимые по проверке зданий и сооружения на соответствие требованиям cGMP, должны в целом продемонстрировать, что здания и сооружения выполнены надлежащим образом и соответствуют предназначенному использованию.
Оборудование. Квалификация оборудования и систем получения технологических сред включает в себя следующую деятельность:
Проверка того, что конструкционные материалы технологического оборудования и систем получения технологических сред, а также их принципы работы и эксплуатационные характеристики, соответствуют требованиям, предъявляемым к ним производством фармацевтической продукции (в общем виде - требованиями GMP, а, в частности, конкретного технологического процесса);
Проверка того, что системы технологических сред и технологическое оборудование надлежащим образом изготовлены, смонтированы, соединены, а средства измерений откалиброваны;
Проверка того, что системы технологических сред и технологическое оборудование функционируют в соответствие с требованиями технологического процесса во всем поле допустимых значений технологических параметров. Эти системы и оборудование следует испытывать под нагрузкой, соответствующей условиям реального производства. Необходимо проверять проведение всех операций, выполняемых на оборудовании (например, запуск в работу, остановка), которые характерны для производственного процесса. Следует продемонстрировать, что поддержание технологических параметров в заданных пределах обеспечивается настолько длительное время, насколько это требуется в реальных производственных условиях.
Работы по квалификации технологического оборудования и систем для получения технологических сред могут проводиться по индивидуальному плану или по плану, являющемуся частью общего плана реализации проекта. Следует заботиться о том, чтобы план содержал требования к выполнению этих работ, а также включал в себя деятельность по управлению рисками для определения приоритетов и распределения усилий как при выполнении, так и при документировании действий по квалификации.
В этот план следует включать следующее:
1) перечень работ или проводимых испытаний;
2) критерии приемлемости для оценки полученных результатов;
3) график проведения квалификационных работ во времени;
4) распределение ответственности за выполнение работ и утверждение их результатов;
5) описание процедур документирования и утверждения результатов квалификации.
В него также следует включать требования предприятия к работам, выполняемым по оценке изменений (управление изменениями).
Все работы по квалификации следует документировать и суммировать в отчете, содержащем заключения, сделанные на основании критериев приемлемости, зафиксированных в плане работ. Отдел контроля качества предприятия должен проверять и утверждать как план, так и отчет по квалификации (21CFR 211.22).
Квалификация эксплуатации (Performance Qualification - PQ).
PQ- является вторым элементом стадии квалификации технологического процесса и проводится на стадии его внедрения в промышленное (коммерческое) производство. PQ выполняется в реальных производственных помещениях, на реальном технологическом оборудовании и с реальными системами технологических сред (каждые из которых предварительно квалифицированы), с персоналом, обученным вести производственный процесс, с использованием реальных процедур управления производственным процессом, а также с использованием реального сырья и материалов, для того, чтобы произвести коммерческие партии готовой продукции. Успешное выполнение PQ должно подтвердить правильность определения технологического пространства процесса (Design Space) и продемонстрировать, что промышленный производственный процесс проводится так, как ожидалось.
Успешное выполнение PQ, показывает, что достигнут важный пункт в жизненном цикле продукции. Т.к. именно успешное выполнение PQ является обязательным условием для начала промышленного производства и коммерческой реализации продукта. Принятие решения о начале коммерческой реализации продукции следует делать на основе данных, полученных при производстве промышленных партий. Данные, полученные на стадиях лабораторных и пилотных исследований, могут дать дополнительное обоснование для принятия такого решения. При принятии решений учитывается общий уровень знаний о процессе, имеющийся на предприятии, а также предыдущий опыт производства аналогичной продукции.
Новое руководство настойчиво рекомендует предприятиям использовать статистические методы для получения статистически достоверной информации. Именно применение статистических методов позволяет определить необходимое и достаточное число партий продукции, которое должно быть наработано при проведении PQ для получения статистически достоверной информации, подтверждающей валидированность процесса.
Во многих случаях PQ предполагает использование большего объема отбора проб и проведения большего числа тестов, чем в обычном производстве. Уровни мониторинга процесса и проводимых испытаний должны быть такими, чтобы подтвердить однообразие качества продукта во всей партии. Предполагается, что таким более высоким уровнем тестирования следует сопровождать всю стадию квалификации процесса.
Обращается внимание на то, что если в процессе производства используются материалы с ограниченным сроком действия (например, фильтры или хроматографические сорбенты), то повторное их использование без потери качества продукта, может быть подтверждено соответствующими лабораторными исследованиями. Однако, при выполнении PQ в коммерческом производстве, следует подтвердить установленные сроки годности таких материалов.
Протокол квалификация эксплуатации ( Protocol PQ).
Для этой стадии валидации процесса необходимо наличие письменного документа (протокола), который определяет требования к условиям производства, мерам контроля, отбору проб и тестированию, а также ожидаемым результатам. Рекомендуется, чтобы этот документ содержал следующее:
Описание условий производства, включая технологические параметры, их предельные значения и требования к сырью и материалам.
Перечень данных, которые собираются при испытаниях, когда и как они оцениваются;
Перечень тестов, которые выполняются в процессе производства и критерии приемлемости для каждого значительного этапа производства;
План отбора проб, включая точки отбора проб, число проб, частоту отбора проб для каждой операции. При этом необходимо, чтобы число проб было достаточным, чтобы получить статистически достоверную информацию о качестве продукции как внутри одной партии, так и сравнительную между партиями;
Критерий для принятия обоснованного заключения о том, что процесс обеспечивает производство продукции надлежащего качества (этот критерий должен включать описание статистических методов, используемых для оценки всех собранных данных, а также порядок учета и обработки данных о выявленных отклонениях);
Данные о проекте производственных помещений, о квалификации технологического оборудования и систем технологических сред, об обучении персонала и о проверке сырья и материалов;
Статус валидации аналитических методов, используемых при измерениях, выполняемых в процессе производства, контроле качества полупродуктов и готовой продукции;
Проверку и утверждение соответствующими службами предприятия и отделом контроля качества.
Выполнение работ по протоколу квалификации эксплуатации и составление отчета.
Работы по протоколу PQ не начинаются до проверки и утверждения протокола PQ соответствующими отделами и отделом контроля качества. Отклонения от протокола PQ, должны быть обоснованы, рассмотрены и утверждены всеми указанными отделами.
Производственный процесс должен выполняться в соответствии с условиями регламента. Партии продукции при PQ следует нарабатывать в нормальных условиях и с тем же производственным персоналом, который будет работать на всех участках производства при коммерческом выпуске продукта. Нормальные производственные условия, должны включать в себя технологические среды (такие как, например, сжатый воздух и вода фармацевтического качества), материалы, персонал, качество производственной среды и производственные процедуры.
После выполнения работ по протоколу PQ следует своевременно составить отчет о выполненных работах. В него необходимо включать:
Обсуждение и рассмотрение всех аспектов протокола;
Итоговый обзор и анализ собранных данных, как это предписывается протоколом;
Оценка любых неожиданных наблюдений и полученных дополнительных данных, которые не были указаны в протоколе;
Обзор и обсуждение всех несоответствий производственного процесса (таких как отклонения и расхождение результатов испытаний) или любой другой информации, способной поставить под сомнение валидированность процесса;
Детальное описание любых корректирующих действий или изменений, которые необходимо было предпринять в существующих технологических операциях и мерах контроля;
Ясное заключение о том, что полученные данные показывают, что процесс отвечает условиям, определенным в протоколе и, что процесс находится в соответствующем состоянии контроля. Это заключение необходимо делать, на основании документированных доказательств для того, чтобы получить разрешение на процесс и на реализацию партий продукции, выпущенной при PQ. Если это не достигнуто, то в отчете следует указать, что необходимо сделать, чтобы достичь такое состояние при повторном PQ.
Включать данные о проверке и утверждении отчета всеми обозначенными отделами и отделом контроля качества.
Стадия 3. Текущая верификация процесса
Цель третьей стадии валидации процесса - получать постоянное подтверждение того, что процесс остается в состоянии контроля (состоянии валидированности) в течение коммерческого производства. Для достижения этой цели следует разработать систему мер для выявления отклонений от процесса. Соответствие требованиям GMP предполагает сбор и анализ информации, которая позволит выявить тенденции, относящиеся к ведению процесса. Этот анализ необходим для своевременного определения того, какие действия следует предпринять, чтобы остановить выход процесса из состояния контроля.
Должна быть разработана система действий по сбору и анализу данных о процессе и продукте. Следует собирать данные о тенденциях, касающихся процесса, а также касающихся качества исходного сырья, полупродуктов и готовой продукции. Эти данные следует статистически обрабатывать для выявления статистически достоверных изменений. Необходимо разработать программу сбора данных о процессе и методику их статистической обработки. Эта методика призвана предохранить от избыточных действий и в тоже время не дать упустить вредные тенденции. Полученная с ее помощью информация должна либо подтверждать стабильность производственного процесса, либо дать своевременный сигнал о необходимости предпринять корректирующие действия, направленные на устранение вредных тенденций.
Такая система действий по сбору и анализу информации о процессе и продукте, призванная либо подтвердить стабильность процесса, либо дать своевременный сигнал о необходимости корректирующих действий в случае появления вредных тенденций и представляет собой текущую верификацию процесса . Эта стадия верификации призвана дать доказательство поддержания процесса в валидированном состоянии в течение всего периода коммерческого производства продукта.
Ремонт и техническое обслуживание помещений, оборудования, калибровка средств измерений, обучение персонала и другие необходимые действия призваны помочь обеспечить стабильность процесса и надлежащее качество готовой продукции.
Валидируемость процесса
Качество продукта и условия обеспечения этого качества в процессе производства, должны быть заложены в продукт на стадии его разработки и на стадии разработки технологического процесса его производства;
Систему обеспечения качества продукции следует создавать с учетом рисков, способных помешать выпуску качественной продукции;
Валидацию процесса, как часть системы обеспечения качества, также следует проводить с учетом анализа рисков, в частности, в условиях «наихудшего случая»;
В целом, новое руководство является последовательным шагом, сделанным FDA в направлении практической реализации нового подхода к cGMP, основанного на анализе рисков.
Литература
- FDA’s “Current Good Manufacturing Practices (cGMP), Guidance for Industry. Process Validation: General Principles and Practices”, November, 2008, Rockville, MD, USA.
- FDA’s “Pharmaceutical cGMPs for the 21 st Century: Risk-Based Approach” Concept Paper, August, 2002, Rockville, MD, USA.
- FDA’s “Current Good Manufacturing Practices (cGMP), Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing”, September, 2004, Rockville, MD, USA.
- FDA’s “Current Good Manufacturing Practices (cGMP), Guidance for Industry. PAT - A Framework for Innovative Pharmaceutical Development Manufacturing and Quality Assurance”, Pharmaceutical cGMPs, September, 2004, Rockville, MD, USA.
- FDA’s “Current Good Manufacturing Practices (cGMP), Guidance for Industry, Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations”, September, 2006, Rockville, MD, USA.
- Попов А.Ю. Валидация критических процессов и зон, «Чистые помещения и технологические среды», № 2, 2005, с. 22-26.
- Попов А.Ю. Валидация и квалификация технологического оборудования, «Чистые помещения и технологические среды», № 2, 2006, с. 38-41.
- Попов А.Ю. Валидация - что, где, когда? «Чистые помещения и технологические среды», № 3 , 2003, с. 34-37.
- ICH Guidance for Industry, Q8A Pharmaceutical Development, May, 2006.
- ICH Draft Guidance for Industry, Q10 Quality Systems, May, 2007.
- Amer Gamal, Corrective Action Preventive Action (CAPA): A Risk Mitigating Quality System, Pharmaceutical Engineering, Volume 28, Number 3, May/June 2008, p.66-72.
- ICH Guidance for Industry, Q9A Quality Risk Management, June, 2006.
- Попов А.Ю., Мешковский А.П. Система анализа риска (HACCP) как первый шаг в переходе к работе по правилам надлежащей производственной практики (GMP), Фарматека, № 4, 2002, стр.62-64.
- Попов А.Ю. Система анализа рисков, Чистые помещения и технологические среды, № 1, 2004, с. 30-32.
Как валидировать процессы производства продукции и насколько важно выделять из них «специальные»?
Качалов В.А.
Журнал «Методы менеджмента качества», 2010, №10-11
Загрузить (размер: 380.3 Кб , скачиваний: 2724 )
Данная публикация - очередная попытка ответить
на «каверзные» вопросы дотошного менеджера по качеству
Речь в настоящей статье пойдет о том, как интерпретировать требования п. 7.5.2 стандарта ISO 9001:2008 .
Автор проанализировал 100 взятых подряд Руководств по качеству компаний, сертифицированных в TUV International Certification, а также нескольких компаний, сертифицированных в других системах сертификации, и установил следующее.
В 57% случаях было заявлено, что требования п. 7.5.2 не применяются, поскольку в компании отсутствуют процессы производства и сервисного обслуживания, результаты осуществления которых не могут быть верифицированы последующим мониторингом или измерениями 1 .
Эти компании осуществляли добычу и переработку руды и каменного угля, производство металла, парфюмерно-косметической продукции, обогащение урана, транспортирование газа, осуществление капитальных ремонтов скважин, обращение с радиоактивными отходами, архитектурно-строительное проектирование, производство изделий из стекла и пластмассы, предоставление услуг по технической диагностике, обслуживанию авиапассажиров, производство строительных конструкций, пищевой продукции, а также диспетчеризацию и снабжение электроэнергией, машиностроение, приборостроение, производство резинотехнических изделий, выполнение строительно-монтажных работ.
В остальных 43% случаях было заявлено о наличии указанных выше процессов и о проведении их валидации в соответствии с требованиями п. 7.5.2. Сюда вошли представители нефтегеофизики, электротехники, автомобилестроения, производства труб, а также диспетчеризации и снабжения электроэнергией, машиностроения, приборостроения, производства резинотехнических изделий и выполнения строительно-монтажных работ.
Таким образом, можно с большой уверенностью говорить, что требования п. 7.5.2 стандарта ISO 9001:2008 применяют от 40 до 50% сертифицированных компаний, а это означает, что в масштабах России речь идет о многих тысячах, если не о десятках тысяч таких компаний.
Вместе с тем, анализ показывает следующее:
Первое. Представители одной и той же отрасли (они выделены выше полужирным шрифтом), применяя практически одни и те же технологии, тем не менее, по-разному оценивают наличие у них процессов, требующих применения требований п. 7.5.2.
Второе. Во многих организациях применяют «собственную» трактовку содержания и смысла действий по валидации процессов, включая требуемую стандартом демонстрацию способности процессов достигать запланированных результатов, например:
Предприятие определило процессы производства как специальные процессы, результаты которых нельзя проверить посредством последовательного мониторинга или измерения. СПОСОБНОСТЬ этих процессов достигать запланированных результатов ПОДТВЕРЖДАЕТСЯ ПОСРЕДСТВОМ применения соответствующего оборудования и квалификации персонала, применения конкретных методов и процедур и ведения соответствующих записей .
Валидация основных процессов... ОБЕСПЕЧИВАЕТСЯ НАЛИЧИЕМ лицензии на право ведения образовательной деятельности, сертификата аттестации образовательного учреждения, свидетельства о государственной аккредитации 2 .
В Руководстве по качеству одной строительной организации раздел «Валидация процессов производства и обслуживания» содержит 8 подразделов, но цитирование их не имеет смысла, поскольку на самом деле ни один из них никакого отношения к действиям по проведению валидации процессов не имеет .
Третье. Чрезвычайно кратки и не всегда понятны разъяснения требований п. 7.5.2 стандарта ISO 9001:2008 в специальной литературе. Вот, например, ПОЛНЫЙ текст комментариев к этому разделу в :
Требования этого подпункта применяют тогда, когда отсутствует возможность измерения параметров и мониторинга процессов в соответствии с подпунктом 8.2.3 «Мониторинг и измерение процессов».
Этот подпункт стандарта относится к ситуациям, когда невозможно оценить выходные результаты процесса или если подобная оценка может быть получена только спустя длительное время.
Организация обязана заранее оценить вероятность возникновения подобных ситуаций, и располагать соответствующими средствами контроля таких процессов.
Никакой дополнительной информации по этому вопросу мы не найдем и в большой монографии . Комментируя обсуждаемые требования, ее автор просто в очень сокращенном варианте пересказал соответствующие требования стандарта ISO 9001 .
Если же говорить о целевых исследованиях, например, о публикации , то они, как будет показано ниже, содержат немало спорных утверждений.
Четвертое. Требования о валидации процессов производства и сервисного обслуживания содержатся не только в стандарте ISO 9001:2008. Они включены также в содержание:
Других международных стандартов, основанных на требованиях стандарта ISO 9001, например, ISO/TS 16949:2009 и ISO/TS 29001:2003 ;
Соглашений, выработанных в ходе международных семинаров (International Workshop Agreement - IWA), например IWA 2:2003 и IWA 4:2005 ;
Международных стандартов, издаваемых другими международными объединениями, например IRIS .
Все эти обстоятельства подтверждают актуальность поиска ответа на вопросы, вынесенные в заголовок статьи.
1. Как валидировать процессы производства продукции
1.1. Требования п. 7.5.2 ISO9001:2008 и «специальные процессы»
Практически все обсуждения требований п. 7.5.2 обычно связаны с понятием специальных процессов, хотя в самом его тексте какие-либо прямые ссылки на эти процессы отсутствуют. Более того, в этом разделе стандарта ISO 9001:2008 и в стандарте ISO 9000:2005, где приведено понятие «специальный процесс», говорится, на первый взгляд, не совсем об одном и том же:
В стандарте ISO 9001:2008 речь идет о процессах, результаты осуществления которых НЕ МОГУТ быть верифицированы последующим мониторингом или измерениями ;
В стандарте ISO 9000:2005 говорится о процессах, в которых верификация соответствия получающейся продукции ЗАТРУДНЕНА или ЭКОНОМИЧЕСКИ НЕЦЕЛЕСООБРАЗНА .
А «не могут» и «затруднено или экономически не целесообразно» - вроде как разные вещи.
На самом деле в обоих источниках говорится, конечно же, об одном и том же. Во-первых, потому, что «затруднено или экономически нецелесообразно», включая их крайний случай «невозможно в настоящих условиях», составляют исчерпывающий перечень причин того, почему «не могут».
Во-вторых (и это - главное), как в первом, так и во втором случае процессов верификация их результатов НЕ ПЛАНИРУЕТСЯ и НЕ ПРОВОДИТСЯ. И именно поэтому недостатки продукции или услуги не могут быть выявлены в ходе или по завершении их создания, а становятся заметными только после того, как продукция начала использоваться или услуга предоставлена .
Вывод первый. Требования п. 7.5.2 распространяются только на те ПРОЦЕССЫ ПРОИЗВОДСТВА ПРОДУКЦИИ, которые в соответствии с терминологий стандарта ISO 9000:2005 относятся к категории «специальных». Ни к каким другим процессам, строго говоря, требования этого раздела никакого отношения не имеют.
Ключевой же особенностью специальных процессов является то, что при их ПРИМЕНЕНИИ получаемые результаты по тем или иным причинам НЕ ПОДВЕРГАЮТСЯ верификации. Это ИСКЛЮЧАЕТ В ПРИНЦИПЕ возможность подтвердить или не подтвердить соответствие данных результатов требованиям, которые относятся к их конкретному предполагаемому использованию или применению, ДО НАЧАЛА их использования или применения.
С учетом этого становится понятной важность требования стандарта ISO 9001:2008 о том, чтобы каждый специальный процесс был валидирован заблаговременно, а именно: еще до своего применения.
1.2. Содержание и цель валидации процессов производства
К сожалению, применение к процессам термина «валидация» в том виде, как он определен в стандарте ISO 9000:2005, а именно: подтверждение (посредством предоставления объективного свидетельства) того, что требования, относящиеся к конкретному предполагаемому использованию или применению, были выполнены, - вызывает вполне определенные затруднения. Помогают преодолеть эту трудность публикации, делающие попытку определить суть валидации процесса 3 , в том числе как:
придания законной силы, утверждения, легализации, ратификации [ 15];
процедуры, дающей высокую степень уверенности в том, что конкретный процесс, метод или система будет последовательно приводить к результатам, отвечающим заранее установленным критериям приемлемости ;
(от лат. valius - сильный, здоровый, соответствующий здоровому) - документированного действия, подтверждающего, что применяемые в исследованиях или производстве методики, процессы, деятельность или системы действительно приводят к ожидаемым результатам в соответствии с нормативной документацией ;
получения документированного доказательства, дающего высокую степень уверенности в том, что процесс будет постоянно производить продукт, отвечающий предварительно установленным требованиям и показателям качества ;
доказательства того, что что-то (процесс и т. д.) работает так, как должно работать. Иными словами, процесс должен работать в соответствии со своим назначением. Для доказательства, что так оно и есть, и служит система последовательно выполняемых проверок и испытаний, называемых валидацией .
Приведенные определения помогают понять, что провести валидацию процесса - значит осуществить контрольные действия для выяснения того, обладает ли разработанный «на бумаге» процесс следующим внутренне присущим ему свойством: при своем применении в точном соответствии с разработанной технологией он позволяет получать результаты, отвечающие требованиям к их конкретному предполагаемому использованию или применению. Если указанные доказательства соответствия будут получены, процесс признается валидированным.
Вывод второй. СОДЕРЖАНИЕМ действий по проведению валидации процесса являются действия по оценке соответствия его результатов требованиям к их конкретному предполагаемому использованию или применению, а ЦЕЛЬЮ валидации - получение уверенности в наличии устойчивой внутренней способности процесса обеспечивать это соответствие при его применении в будущем.
1.3. Валидация процессов производства и валидация продукции
Валидация процесса требует проведения действий по оцениванию соответствия его результатов (произведенной продукции или полуфабриката) неким требованиям. А это, говоря языком стандарта ISO 9000:2005, - действия по верификации . При этом нетрудно заметить: если при проведении ВЕРИФИКАЦИИ произведенной продукции в состав установленных требований будут включены требования, относящиеся к конкретному предполагаемому использованию или применению, то такая верификация автоматически становится ВАЛИДАЦИЕЙ продукции .
Это означает, что: когда в критерии приемки производимой с помощью какого-то процесса продукции (в том числе промежуточной) включены требования, относящиеся к ее конкретному предполагаемому использованию или применению, а по завершении процесса МОЖНО провести и ПРОВОДЯТ верификацию произведенной продукции на соответствие таким критериям приемки, то положительные результаты данной верификации одновременно будут доказательствами валидации и самой продукции, и процесса ее производства. Более того, именно на этом принципе и построен обычно применяемый механизм признания, утверждения и т. д. (в конечном счете - валидации) производственных процессов - на валидации произведенной с их помощью продукции 4 .
Другое дело, если полученную продукцию по тем или иным причинам НЕЛЬЗЯ подвергнуть процедуре верификации. Как быть в этом случае? Это действительно вопрос, притом - самый важный для этой части статьи. Именно об этом пойдет речь ниже.
Вывод третий. Положительные результаты валидации продукции являются прямым доказательством валидированности (принципиальной приемлемости для своего применения) процесса ее производства, ибо тем самым они демонстрируют наличие у этого процесса внутренней способности получать то, что от него требовалось.
1.4. Валидация специального процесса и разрешение на его конкретное применение
Специальный процесс, как и любой другой, разрабатывают для его последующего ПРИМЕНЕНИЯ. Однако начинать его реализацию вряд ли целесообразно, если у него нет ДВУХ последовательно полученных разрешений на применение - базового и частного. Именно на это указывает содержание п. 7.5.2.
1.4.1. Базовое разрешение на применение
Его наличия требует первый абзац этого раздела стандарта, в котором говорится о необходимости валидации специальных процессов. Поясним смысл данного утверждения.
Валидация, как сказано выше, подтверждает наличие у процесса ВНУТРЕННЕЙ СПОСОБНОСТИ достигать желаемых результатов. При этом данное подтверждение должно базироваться на демонстрации способности процесса достигать запланированных результатов, опирающейся на установленные критерии. Поэтому автор разделяет точку зрения тех, кто заявляет: валидация процесса является основанием для его ОФИЦИАЛЬНОГО утверждения .
При этом исключительно важно понимать, ЧТО ИМЕННО происходит в ходе действий по валидации процесса производства.
Приобретение этого статуса возможно лишь тогда, когда при реализации этого процесса:
а) ИСПОЛНИТЕЛЯМИ работ, чья компетентность была не ниже требований, установленных в описании разработанного процесса,
б) с помощью ОБОРУДОВАНИЯ, имеющего характеристики не хуже тех, что установлены в описании разработанного процесса,
в) из СЫРЬЯ, МАТЕРИАЛОВ, КОМПЛЕКТУЮЩИХ, имеющих характеристики не хуже тех, что установлены в описании разработанного процесса,
г) в ПРОИЗВОДСТВЕННОЙ СРЕДЕ, характеристики которой не хуже тех, что установлены в описании разработанного процесса, и
д) в точном соответствии с ТЕХНОЛОГИЧЕСКИМИ ХАРАКТЕРИСТИКАМИ ОПЕРАЦИЙ (температура, сила тока, химический состав и т. д.), которые установлены в описании разработанного процесса БЫЛА ПОЛУЧЕНА ПРОДУКЦИЯ (в том числе промежуточная), соответствующая требованиям, которые относятся к ее конкретному предполагаемому использованию или применению. Другими словами, хотя бы раз в ТАКИХ условиях своего применения процесс действительно дал «на выходе» то, что от него хотели.
Применение к процессу дополнений «валидирован», «санкционирован», «аттестован» или «утвержден» означает, что, В ТОМ ВИДЕ, в котором его представили разработчики, он хотя бы раз прошел процедуру валидации с положительными итогами, на этом основании официально признан приемлемым и допущен к своему применению в будущем. Валидация сигнализирует, что у процесса имеется базовое (исходное, основополагающее) разрешение на применение или разрешение на применение процесса как ТАКОВОГО 5 . Он В ПРИНЦИПЕ готов к применению.
Но, заметим особо, И ТОЛЬКО.
1.4.2. Частное разрешение на применение
Сам по себе конкретный «запуск в производство» специального процесса, даже валидированного, еще никак не означает, что мы по его окончании получим то, что хотелось.
Если нужно с большой долей уверенности получить от применения валидированного процесса те же результаты, на которые он потенциально способен и которые он «выдал» при своей валидации, необходимо в КАЖДОМ конкретном случае обеспечить наличие таких условий его реализации, которые являлись бы НЕ ХУЖЕ тех, что были при его валидации. А именно: обеспечить соответствие компетентности участников работ, применяемого оборудования, сырья, материалов, комплектующих, производственной среды и технологических параметров осуществляемых операций требованиям, установленным в документации валидированного процесса. В теории менеджмента качества эти составляющие известны как пять «М» (Man, Mechanism или Machine, Material, Milieu, Method).
Получение частного разрешения на применение и означает, что у валидированного процесса имеются в наличии все необходимые «М» и он тем самым ПОДГОТОВЛЕН к началу своей конкретной (отдельно взятой, частной) реализации с ДОСТАТОЧНОЙ уверенностью в положительном исходе.
Именно об этом говорится в третьем абзаце п. 7.5.2, где требуется, чтобы организация:
одобрила (признала приемлемость) оборудования, которое планируют применять в данном процессе, и квалификацию персонала, который будет участвовать в реализации данного процесса;
применила специфические (предназначенные именно для этого процесса) методы и процедуры, т. е. именно те, которые были валидированы и имеют базовое разрешение на применение;
Установила записи, подтверждающие наличие как всех исходных (валидированная технология, приемлемые оборудование, персонал, сырье, материалы и комплектующие), так и обеспечивающих компонентов процесса (соблюдение технологии и требований к производственной среде 6).
Важно подчеркнуть, что частное разрешение на применение является САМОСТОЯТЕЛЬНЫМ необходимым разрешением на КОНКРЕТНОЕ применение процесса. Оно не только не отменяет, а ПРЕДПОЛАГАЕТ наличие обозначающего валидированность процесса в целом. Вместе с тем, анализ Руководств по качеству показал, что разницу между базовым и частным разрешением на применение признают и учитывают далеко не все.
Например, в одном из Руководств по качеству утверждается: СПОСОБНОСТЬ этих процессов достигать запланированных результатов подтверждается ПОСРЕДСТВОМ ПРИМЕНЕНИЯ соответствующего оборудования и квалификации персонала, применения конкретных методов и процедур и ведения соответствующих записей.
В другом сказано, что ВАЛИДАЦИЯ специальных процессов ВКЛЮЧАЕТ:
аттестацию используемого оборудования;
исследование применяемых материалов;
подготовку и аттестацию персонала.
В третьем, что КРИТЕРИЯМИ ДЛЯ УТВЕРЖДЕНИЯ [в смысле валидации - прим. авт.] процесса ЯВЛЯЮТСЯ:
результаты аттестации персонала;
результаты аттестации оборудования;
выполнение графика контроля технологической дисциплины и его результаты.
Как видно, в этих случаях, говоря о действиях по проведению валидации процесса, т. е. по получению базового разрешения на применение, организации на самом деле вкладывают в них смысл действий по получению
Вывод четвертый. Выражение «валидированный процесс», конечно же, означает, что данный процесс «утвержден», «аттестован», «санкционирован/разрешен для применения» и т. п., поскольку продемонстрировал хотя бы раз соответствие КРИТЕРИЮ ВАЛИДАЦИИ - свою способность достигать запланированных результатов. Но это - лишь утверждение процесса «в принципе», означающее наличие у него «БАЗОВОГО разрешения на применение».
Для успеха при КОНКРЕТНОМ применении валидированного процесса требуется обеспечить наличие еще и «ЧАСТНОГО разрешения на применение». Основанием для его получения будет соответствие процесса ДРУГИМ КРИТЕРИЯМ, а именно: обеспечивающим уверенность, что этот процесс при данном конкретном применении будет осуществляться в тех же условиях, что и при своей валидации.
Для гарантированного выполнения любого конкретного заказа у применяемых специальных процессов должны быть ОБА указанных разрешения на применение.
1.5. Ошибки при признании специальных процессов валидированными
Анализ Руководств по качеству и публикаций показал, что во многих случаях требуемая в п. 7.5.2 валидация специальных процессов производства, т. е. получение базового разрешения на применение, вольно или невольно, но ПОДМЕНЯЕТСЯ или даже ЗАМЕНЯЕТСЯ получением частного разрешения на применение. Характерным примером этого является методика валидации процессов одной из организаций, описанная в публикации .
В ней обоснованно заявлено, что для валидации/ утверждения специального процесса нужны критерии/требования, и что за 10 дней до даты валидации составляются протоколы подтверждения соответствия факторов установленным требованиям, на основании чего комиссия выносит решение об утверждении специального процесса либо о необходимости его повторной валидации.
И все бы ничего, но оцениваемыми факторами здесь были выбраны не характеристики продукции (а организация предоставляет услуги связи), и не степень соответствия их требованиям, относящимся к конкретному предполагаемому использованию или применению. Анализируемыми факторами были: оборудование, персонал, методики, программное обеспечение, производственная среда и ресурсы. И проверялись они на соответствие тому, что было установлено В ПРОЦЕДУРАХ оказания услуг связи. А это означает, что ФАКТИЧЕСКИ осуществлялись действия по получению не базового, а частного разрешения на применение.
Поэтому, если строго следовать описанной в методике, то принимаемое в этой организации реше ние об утверждении специального процесса (т. е. решение о валидации процесса) на самом деле никакого отношения к ВАЛИДАЦИИ не имеет, поскольку базируется исключительно на оценке соответствия тех или иных «М» требованиям, установленным в описании процесса, а не на оценке соответствия РЕЗУЛЬТАТОВ процесса требованиям к этим результатам. А вот были ли валидированы соответствующие ПРОЦЕДУРЫ (т. е. СОДЕРЖАНИЕ) процесса и каким именно образом - эти вопросы остались в данной публикации без ответа.
Вывод пятый. Нередки случаи, когда в организациях валидацию процесса, названного специальным, требующую получения свидетельств соответствия его результатов требованиям, относящимся к конкретному предполагаемому использованию или применению, заменяют действиями по получению свидетельств выполнения УСТАНОВЛЕННЫХ разработчиками этого процесса требований к исполнителям, сырью, оборудованию, производственной среде и т. п., часто называя это «утверждением» или «аттестацией» специального процесса. При этом наличие приемлемости (валидированности) самого процесса не анализируется, по умолчанию считается доказанной когда-то в прошлом, ничем не подкрепляется, а подтверждение этой валидированности в будущем (т. е. перевалидация) никак не планируется.
Важно понимать, что при сертификации подобный подход должен быть оценен аудиторами как несоответствие требованиям первого абзаца и последнего подпункта третьего абзаца п. 7.5.2 стандарта ISO 9001:2008.
Справедливости ради следует отметить, что в некоторых организациях, определяя понятия «валидация», «утверждение» или «аттестация» специальных процессов, учитывают необходимость наличия ОБОИХ рассмотренных выше обстоятельств, что является, конечно же, правильным. Сюда же надо добавить и тех специалистов, которые в своих публикациях, определяя понятие «валидация процесса», также СРАЗУ объединяют в «один пакет» базовое и частное разрешение на применение, говоря о валидации, например, как о документированном подтверждении со ответствия оборудования, условий производства, технологического процесса, качества полупродукта и готового продукта действующим регламентам и/или требованиям нормативной документации . Однако, к сожалению, выборочный анализ показал, что подобные организации и публикации - скорее исключение, чем правило.
1.6. Как валидировать специальные процессы?
В формулировках стандарта ISO 9001:2008 валидация означает, что организация должна КАКИМ-ТО образом продемонстрировать способность специальных процессов достигать запланированных результатов. В том, как это можно сделать, и содержится искомый ответ.
К сожалению, в большинстве проанализированных «Руководств...» самым распространенным был вариант, когда в тексте без каких-либо дополнительных комментариев или ссылок на какие-либо процедуры просто констатировалось, что валидация процессов про изводства продукции демонстрирует способность процессов достигать запланированных результатов. И все! Вопрос о том, КАК ИМЕННО была продемонстрирована эта способность, остался там без ответа. А ведь это - самое важное.
Представляется, что указанные выше особенности специальных процессов позволяют говорить о двух методах их валидации.
1.6.1. Валидация на основе оценки получаемой продукции
Напомним, что к специальным процессам относятся, В ТОМ ЧИСЛЕ, те, для которых верификация соответствия получающейся продукции ЗАТРУДНЕНА. Однако заметим, это не означает, что она В ПРИНЦИПЕ НЕВОЗМОЖНА. Более того, из-за того, что такая верификация для целого ряда специальных процессов в принципе технически возможна, именно этот прием в большинстве случаев и используется для их валидации.
Вот характерный пример из Руководства по качеству, описывающий способ валидации специального процесса: через определенные технической документаци ей промежутки времени характеристики продукции подвергаются инструментальному контролю в целях валидации процесса и верификации продукта этого процесса.
Вместе с тем, внимательный читатель скажет, что этот способ валидации процессов производства уже описан выше и даже назван там обычно применяемым. Поэтому вопрос заключается в том, существуют ли какие-то отличия в применении данного метода по отношению к специальным процессам?
Ответ следующий: никаких методических отличий между использованием этого способа валидации применительно к обычным и специальным процессам производства нет. Однако есть ОСОБЕННОСТИ. Они заключаются в том, что обычно (так уж получается) организовать и провести верификацию результатов специальных процессов гораздо ТРУДНЕЕ, чем результатов других процессов.
Например, в принципе МОЖНО проверить результаты процесса нанесения защитного гальванического покрытия на изделия, предназначенные для работы в экстремальных условиях. Но для этого нужно будет «всего лишь» поместить несколько образцов в соответствующие условия (напрямую или создать их искусственно), а после определенного срока применить разрушающие методы контроля, требующие специального оборудования и программных средств, подготовленного персонала и т. д. Согласитесь, это намного труднее организовать и сделать, чем, например, проанализировать соответствие геометрических характеристик изделий после их механической обработки с помощью типовых средств линейно-угловых измерений.
И тем не менее, главное здесь другое: хоть и редко, но организации В ПРИНЦИПЕ могут позволить себе это делать. И делают.
Так, в одном из Руководств по качеству указано, что валидация процессов основана на результатах приемо-сдаточных контроля и испытаний опытной партии продукции, изготовленной:
а) в отсутствие замечаний по состоянию и работе оборудования, задействованного в аттестуемом процессе производства;
б) в отсутствие задействованных в аттестуемом процессе производства контрольно-измерительных приборов, не прошедших поверку (калибровку) или с просроченным сроком поверки (калибровки);
в) технологическим персоналом, который имеет необходимую квалификацию;
г) в условиях, когда все технологические параметры производства продукции соответствуют требованиям, установленным в аттестуемом описании процесса.
В этой компании процесс считается валидированным, если изготовленная в указанных условиях продукция полностью соответствует требованиям, установленным в нормативно-технической документации.
А вот что касается повторной валидации (перевалидации) специальных процессов, о которой говорится в п. 7.5.2, то, с учетом указанных проблем, рассматриваемый способ вряд ли позволит делать это часто. Во всяком случае, понятно, что проверку соответствия геометрическим размерам можно позволить себе делать намного чаще, чем проверку характеристик защитного покрытия. Нет сомнения, что разработчики стандарта ISO 9001:2008 именно это обстоятельство имели в виду, позволяя организации самой установить либо периодичность, либо конкретные даты следующих действий по повторной валидации (перевалидации) специальных процессов.
В общем же случае критериями выбора интервала или времени следующей плановой валидации специального процесса является разумный баланс между важностью поддержания его статуса валидированного и возможностями выделить на повторную валидацию соответствующие трудовые, материальные, финансовые и другие ресурсы. С учетом этого в одной из компаний установлено, что перевалидация специальных процессов проводится через год, а также в течение первых двух месяцев после проведения капитального ремонта оборудования, участвующего в реализации этого процесса. В другой же компании повторная валидация осуществляется только через пять лет.
Вывод шестой. Валидация специальных процессов производства на основе верификации полученной продукции методически ничем не отличается от валидации других процессов. Вместе с тем, возможность выделения ресурсов для валидации, а затем и для перевалидации специальных процессов обычно является серьезной проблемой, поскольку верификация результатов специальных процессов обычно более сложна и трудоемка, чем результатов обычных процессов производства.
1.6.2. Валидация на основе прогнозных оценок
Хорошо, если верификация результатов специального процесса в принципе возможна, пусть это и вызывает дополнительные сложности. Но ведь могут быть случаи, когда это НЕВОЗМОЖНО - по техническим причинам, из-за временных ограничений или по экономическим соображениям.
Примерами, связанными с техническими и временными обстоятельствами, могут быть процессы создания корпуса ядерного реактора, который должен выдержать колоссальную радиационную нагрузку в течение 30 лет, не потеряв при этом своих прочностных характеристик. Чтобы верифицировать эти характеристики в полном объеме, надо загрузить в созданный корпус настоящий реактор, обеспечить, чтобы он проработал 30 лет, и лишь после этого измерить интересуемые прочностные характеристики. Очевидно, что это просто невозможно - ни технически, ни по объему требуемого времени.
Примером экономических ограничений может служить случай, когда для валидации процесса необходимо фактически уничтожить созданное изделие, а оно очень дорогое и заказано всего в одном или нескольких экземплярах. Понятно, что в этих условиях изготовление еще одного изделия ИСКЛЮЧИТЕЛЬНО для валидации процесса его производства, которое при этом будет уничтожено, вряд ли экономически обоснованно.
В таких случаях более разумно обсуждать и рассматривать лишь «гипотетический» результат специального процесса. Вместе с тем, и здесь с той или иной погрешностью можно проанализировать приемлемость различных прогнозных вариантов этих результатов, получаемых на основе применения ряда известных методов: обобщения экспертных точек зрения, аналогий, расчетно-экспериментальных экстраполяции, моделирования хода самих процессов и условий последующего применения или использования получаемой продукции и т. д.
Конечно, здесь неопределенность положительных заключений о валидации гораздо выше, чем при прямой верификации «живых» результатов процесса. Поэтому при появлении любой новой научно-технической информации и/или нового опыта, относящихся к методам осуществления валидации на основе прогнозов, следует сразу же проводить анализ адекватности примененных ранее методов и, в зависимости от итогов анализа, решать вопрос о повторной валидации с учетом ставших доступными новых знаний.
Вывод седьмой. В отсутствии возможности проведения валидации специальных процессов на основе прямой верификации, созданной на их основе продукции, их валидацию следует проводить, используя методы прогнозирования .
Ввиду очевидно более высокой неопределенности получаемых при этом оценок, организации следует рассматривать вопрос о необходимости и/или целесообразности перевалидации специальных процессов всегда, как только становятся известными новые знания о прогнозных методах, применяемых для валидации этих процессов.
1.7. Все ли процессы, названные «специальными», являются таковыми?
Ответ на вопрос о том, что такое «специальный процесс», дан выше. Однако эту точку зрения разделяют далеко не все.
Например, в публикации к специальным процессам отнесены процессы, результаты которых будут скрыты при выполнении последующих процессов (стадий, этапов). Однако уже далее авторы, говоря о получении объективных свидетельств, необходимых для валидации ТАКИХ процессов, относят к свидетельствам результаты ПРИЕМКИ и/или ОСВИДЕТЕЛЬСТВОВАНИЯ скрытых работ, отдельно предъявляемых операций, операционного контроля. Это означает, что верификация результатов этих процессов не только возможна, но и ПРОВОДИТСЯ, притом - фактически ВСЕГДА. А это никак не подпадает под признаки специальных процессов, описанные в стандарте ISO 9000:2005.
Немалая часть споров связана с наличием или отсутствием специальных процессов при предоставлении услуг. Например, авторы относят к специальным процессы, которые включают предоставление услуг в реальном масштабе времени и выполняются непосредственно при контакте с потребителем (например, регистрация прибытия и выписка клиента в отеле или обеспечение ремонта продукции по месту нахождения клиента).
Аналогично, говоря о процессе предоставления образовательных услуг, авторы не ставят ни под какое сомнение тот факт, что образовательная деятельность представляет собой процесс, результаты которого не могут быть верифицированы последующим мониторингом или измерениями.
Автор считает, что специальные процессы ИЗНАЧАЛЬНО входят в состав лишь тех услуг, которые оказываются ОЧНО. Что касается услуг, оказываемых ЗАОЧНО, то здесь в большинстве случаев специальных процессов нет, о чем автор уже высказывался ранее в публикации . Поэтому автор разделяет мнение, высказанное в публикациях , которые считают таковыми процессы, относящиеся к контролю и испытаниям (рентгено графия, цветная дефектоскопия, ультразвуковой контроль, испытания под давлением и др.). И м слово в слово вторят авторы , а также поддерживают авторы , заявляя, что методы испытания продукции относятся к центральным объектам валидации.
И здесь автор готов поспорить, считая, что к специальным следует относить лишь те процессы контроля, которые основаны на разрушении/повреждении исследуемого изделия, или которые невозможно повторно применять для одной и той же продукции из-за чрезвычайно высокой скорости ее «старения».
Во всех же других случаях РЕЗУЛЬТАТЫ контроля МОГУТ быть верифицированы либо повторением тех же контрольных операций, либо с помощью ДРУГИХ методов контроля. И, значит, они не попадают под определение «специального процесса».
Есть много и других примеров идентификации каких-то процессов как специальных одними авторами и несогласие с ними других, включая примеры, приведенные в самом начале статьи.
Вывод восьмой. Вопрос о том, является ли какой-то конкретный процесс специальным или нет, очень часто вызывает споры и наличие прямо противоположных точек зрения.
Вместе с тем, представляется, что подобные споры ничтожны, поскольку на самом деле никакого ОСОБОГО ЗНАЧЕНИЯ то, относят тот или иной процесс производства продукции к специальным или нет, вообще говоря, НЕ ИМЕЕТ.
2. Насколько важно выделять из процессов производства продукции «специальные процессы»?
2.1. Валидация процессов производства продукции: только ли «специальных»?
Если мы внимательно проанализируем требования других разделов стандарта ISO 9001:2008, в частности п. 7.1, то увидим, что в стандарте ФАКТИЧЕСКИ подразумевается, чтобы валидированными, т. е. получившими базовое разрешение на применение, были не только специальные процессы, а фактически ВСЕ процессы производства продукции. Давайте проанализируем следующие три стандартные ситуации.
а) Если организация применяет ТИПОВЫЕ производственные операции/процессы, то они являются валидированными по определению, поскольку признание их типовыми невозможно без их соответствующей валидации.
б) Если организация для производства продукции использует процессы, ЗАИМСТВОВАННЫЕ в других организациях, то разумно считать, что никакое заимствование организация не может считать для себя возможным до тех пор, пока не получит доказательств валидированности этих процессов.
в) Наиболее сложной является ситуация, когда процессы производства продукции СОЗДАЮТСЯ в самой организации - в соответствии с подпунктом б) п. 7.1. Заметим, однако, что в этом случае в стандарте требуется, чтобы организация:
1) установила относящиеся к продукции требования, включая критерии ее приемки. И было бы совершенно алогично считать, что в число этих требований НЕ ВОЙДУТ те, которые относятся к ее конкретному предполагаемому использованию или применению - как по отношению к конечной продукции, так и по отношению к ее промежуточным состояниям или полуфабрикатам;
разработала процессы, необходимые для создания продукции (в том числе, конечно, именно процессы ПРОИЗВОДСТВА продукции, а не только процессы проектирования, закупок, контроля и т. п.), а также установила записи, необходимые для предоставления доказательств того, что процессы создания продукции отвечают требованиям . И здесь было бы алогично считать, что в число этих последних требований НЕ ВОЙДЕТ требование обеспечения соответствия получаемой продукции указанным выше критериям приемки.
Поэтому организация будет считать приемлемым для себя лишь такой разработанный производственный процесс, который обеспечит соответствие получаемой с его помощью продукции требованиям к ее конкретному предполагаемому использованию или применению. А это означает, что он ДОЛЖЕН быть соответствующим образом валидирован.
С учетом вышесказанного в любой организации, действующей по модели стандарта ISO 9001:2008, все процессы производства продукции изначально СЛЕДОВАЛО БЫ относить к группе валидируемых 8 . По этой причине нельзя считать обоснованным заявление авторов |7, с. 33] о том, что критерием отнесе ния любых процессов организации к ВАПИДИРУЕМЫМ является ТОЛЬКО то, что их нельзя верифицировать последующим мониторингом и измерениями.
Вывод девятый. Хоть это и не заявлено напрямую, но в модели стандарта ISO 9001:2008 по своей СУТИ предполагается, что процедуре валидации должны быть подвергнуты ВСЕ применяемые процессы производства продукции. А в отношении тех процессов производства, которые разрабатывает сама организация, это, фактически, является прямым требованием.
2.2. Требует ли разд. 7.5.2 валидации других процессов СМК?
Если исходить из логики менеджмента качества, то валидация необходима не только всем процессам производства продукции, но и вообще ЛЮБОМУ процессу, который в п. 4.1 стандарта ISO 9001:2008 назван необходимым для СМК. В противном случае не будет никаких гарантий, что в ходе реализации невалидированного процесса его «выход» будет соответствовать желаемому результату.
На это обстоятельство обращают внимание и другие авторы, отмечая: философия качества гласит, что самый эффективный по стоимости способ ведения бизнеса - это приложение философии специальных процессов ко всем процессам организации . Автор полностью разделяет эту точку зрения.
Вместе с тем, еще раз особо отметим, что в стандарте ISO 9001:2008 требование о необходимости валидации процессов напрямую присутствует только в п. 7.5.2 и отнесено ТОЛЬКО к процессам производства продукции. Это означает, что, строго говоря, требование о валидации не распространяется ни на какие другие процессы, кроме специальных процессов производства продукции. В том числе это требование не относится к процессам проектирования и разработки, хотя на последние часто ссылаются, обсуждая проблемы толкования термина «валидация».
Вывод десятый. Требования п. 7.5.2 стандарта ISO 9001:2008 относятся исключительно к специальным процессам производства продукции и ее сервисного обслуживания и не распространяются ни на какие другие процессы, необходимые для СМК.
2.3. Насколько важно не ошибиться в отнесении производственного процесса к «специальным»?
Понятно, что если какой-то процесс производства продукции НЕОБОСНОВАННО отнесли к специальным, это будет методической ошибкой. Но, с точки зрения целей менеджмента качества, это - «ошибка в лучшую сторону». Поскольку вслед за этим на такой процесс должны будут распространить требования п. 7.5.2 стандарта ISO 9001:2008, а именно: его должны будут ВАЛИДИРОВАТЬ и начинать реализацию только после получения частного разрешения на применение. А это для организации, вне сомнения, хорошо.
Зато совсем другое дело, когда процесс по своей сути ЯВЛЯЕТСЯ специальным, а его не идентифицировали в этом качестве и поэтому не распространили на него требования п. 7.5.2 со всеми вытекающими последствиями. Однако, как представляется, опасения относительно катастрофичности этих последствий «сильно преувеличены».
Из того, что сказано выше, читатель должен вынести, что ЛЮБОЙ процесс производства продукции фактически должен быть до своего применения валидирован (что в «нормальных» компаниях на практике и происходит), а затем осуществляться в управляемых условиях, содержание которых указано в п. 7.5.1. А это означает, что для этих процессов, если они соответствуют требованиям пп. 7.1 и 7.5.1, все требования п. 7.5.2 будут выполняться АВТОМАТИЧЕСКИ, кроме всего одного пункта - о перевалидации.
Вывод одиннадцатый. Если организация В ПОЛНОМ ОБЪЕМЕ выполняет требования пп. 7.1 и 7.5.1 стандарта ISO 9001:2008, то неотнесение каких-то процессов к категории «специальных» не влечет за собой автоматически невыполнение требований п. 7.5.2. Модель стандарта ISO 9001:2008 ФАКТИЧЕСКИ заставляет организацию все равно и валидировать такие процессы, и применять к ним все другие требования п. 7.5.2 (за исключением, возможно, требований о перевалидации), даже не делая на это прямых ссылок.
Из этого вытекает очень важное для организаций следствие. Если в ходе сертификации аудиторы придут к заключению, что какой-то процесс по своей сути является специальным, но организация не при дала ему этот статус, это НЕ ДОЛЖНО автоматически приводить к констатации несоответствия требованиям п. 7.5.2. Вполне возможно, что ФАКТИЧЕСКИ все требования этого раздела в данной организации выполняются в ходе реализаций требований пп. 7.1 и 7.5.1. Аудиторы обязаны выяснить это ДОПОЛНИТЕЛЬНО, чтобы принимаемое ими решение о соответствии или несоответствии требованиям п. 7.5.2 было в полной мере обоснованным.
И еще один вопрос.
Как быть, если в организации при анализе процессов производства будет выявлено, что в ней уже ДАВНО используются какие-то процессы, которые (если опираться на стандарт ISO 9000:2005) должны были быть идентифицированы как «специальные», но которые ранее НЕ УПРАВЛЯЛИСЬ в полной мере так, как того требуется в п. 7.5.2 стандарта ISO 9001:2008?
Ответ здесь однозначен: организация должна НАЧАТЬ управлять этими процессами в соответствии с требованиями п. 7.5.2, начиная с проведения их ОФИЦИАЛЬНОЙ валидации/перевалидации.
Вывод двенадцатый. Во всех случаях в любой сертифицированной организации целесообразно периодически (например, раз в год) проводить анализ своих процессов производства продукции, чтобы уточнить те из них, которые соответствуют определению «специальных», и распространить на них требования п. 7.5.2 стандарта ISO 9001:2008.
2.4. Так нужно ли выделять среди процессов производства «специальные»?
С учетом реальной практики применения в СМ К требований пп. 7.1 и 7.5.2 и опираясь на аргумент полезности, автор без колебаний дает на этот вопрос положительный ответ. Более того, он считал бы рациональным ВСЕ процессы производства признавать «специальными». Но с одной оговоркой. Признавая какой-то процесс «специальным» и/или «особо важным», организация должна вслед за этим:
1. УСТАНОВИТЬ методику его валидации, ДОБИТЬСЯ его валидации и УСТАНОВИТЬ режим перевалидации. Тем самым она может говорить о наличии базового разрешения на применение этого процесса.
2. Перед его каждым конкретным применением осуществлять действия по получению частного разрешения на это применение на основе УСТАНОВЛЕННЫХ КРИТЕРИЕВ проведения анализа и призна ния приемлемости процесса к применению, в число которых должно войти, как минимум, признание при емлемости запланированного к применению оборудования и задействованного персонала.
В ходе реализации процесса обеспечить надзор за тем, чтобы в его ходе применялись только предназначенные ИМЕННО ДЛЯ ЭТОГО ПРОЦЕССА методы и процедуры, а также ВЕЛИСЬ ЗАПИСИ, подтвержда ющие как выполнение критериев приемлемости процесса, так и соблюдение установленных методов и процедур. Вывод тринадцатый. Круг процессов производства, которые организация посчитает для себя критически или особо важными, может выходить за рамки процессов, являющихся специальными. Отнесение процесса к числу критически важных предполагает особое внимание к его подготовке и реализации. Наилучшим механизмом для реализации этого является распространение на такие процессы требований п. 7.5.2 стандарта ISO 9001:2008.
Заключение
Идеологическое. Появление в тексте стандарта ISO 9001:2008 п. 7.5.2 подчеркивает важность внимания, которое Международная организация по стандартизации считает необходимым уделять в организациях специальным процессам из-за связанного с ними повышенного риска для менеджмента качества, обусловленного их спецификой.
Несмотря на то что правила управления процессами производства, сформулированные в пп. 7.1 и 7.5.1, по своей сути в совокупности те же, что и в п. 7.5.2, выполнение требований последнего раздела позволяет организации в гораздо большей степени быть уверенной в минимизации рисков, связанных с возможным непониманием особенностей управления именно такими процессами. С учетом внутренне присущей специальным процессам «критичности», подобный подход представляется полностью оправданным.
Повышенное внимание, которого требует стандарт в отношении специальных процессов, логично распространить и на все другие критически важные процессы.
Методическое. При прохождении сертификации в отношении каждого применяемого специального процесса необходимо продемонстрировать наличие совокупности двух разрешений на их применение. Первое (базовое) - в виде доказательств валидированности процесса, т. е. доказательств его способности в принципе достигать запланированных результатов.
Второе (частное) - в виде подтверждения подготовленности процесса к его конкретному применению. Обычно это происходит в результате разработки и реализации планов технической подготовки соответствующего производства. Критерием же наличия этого разрешения является выполнение требований, установленных в описании процесса ко всем входящим в процесс компонентам, а именно: к исполнителям, оборудованию, исходным материалам, производственной среде и т. д.
Ни одно из этих двух разрешений не заменяет другого. Они должны быть в наличии оба.
Практическое. Если специальный процесс имеет оба указанных выше разрешения на применение, степень уверенности в получении желаемого результата будет зависеть в дальнейшем только от одного - насколько точно соблюдаются установленные разработчиками технологические режимы. Для получения уверенности в этом организация должна установить и обеспечить ведение соответствующих записей.
Представляется, что именно в этих трех обстоятельствах содержится ключевой смысл требований п. 7.5.2 стандарта ISO 9001:2008, которые автор рекомендует любой организации распространить и на другие процессы, важные для СМК.
1 Здесь и далее цитаты из документов выделены полужирным кур-сивным шрифтом. Выделение слов и фраз прописными (заглавными) буквами сделано автором. Цитирование отдельных Руководств по ка-честву со ссылкой на разработавшие их компании сделано только в отдельных случаях.
2 Цитируемое положение было взято из Руководства по качеству вуза, которое ранее было размещено на его сайте, но к моменту под-готовки статьи к опубликованию по каким-то причинам оттуда было изъято.
3 Хотя есть и довольно странные исключения. Например, начав-шийся печататься в специализированном журнале по вопросам ме-неджмента качества «Терминологический словарь системы тех-нического регулирования» не содержит в себе термина «валидация».
4 Следует отметить, что на постсоветском пространстве в ходу остал-ся термин «аттестация процессов». Его часто используют ВЗАМЕН тер-мина «валидация». Проблема в том, что в одних случаях он действитель-но является эквивалентом валидации, например когда аттестацию про-цессов производства определяют в Руководстве по качеству как докумен-тальное подтверждение того, что процесс, протекающий в пределах уста-новленных параметров, обеспечивает результативное, эффективное и воспроизводимое производство ПРОДУКЦИИ, УДОВЛЕТВОРЯЮ-ЩЕЙ УСТАНОВЛЕННЫМ ТРЕБОВАНИЯМ и нормативам качества. Зато в других Руководствах по качеству содержание действий, назы-ваемых «аттестацией процесса», никак нельзя отнести к его валида-ции. Подробнее об этом говорится в последующих разделах статьи.
5 Естественно, мы не рассматриваем здесь ситуацию, когда, по тем или иным причинам, ВНЕ логики менеджмента качества, какое-то лицо силой данных ему полномочий разрешает применять невалидированный процесс. В подобных случаях надо говорить не о «разреше-нии», а о «приказе».
6 Строго говоря, В п. 7.5.2 ничего не говорится о материалах, сырье и комплектующих, равно как и о производственной среде. Однако это, как представляется, чисто «текстуальная» недоработка данного раздела, поскольку эти требования содержатся в других разделах стандарта, ТАК-ЖЕ применимых к специальным процессам, - пп. 7.4 и 6.4.
7 Строго говоря, в п. 7.5.2 говорится ТОЛЬКО о процессах ПРО-ИЗВОДСТВА продукции, а процессы КОНТРОЛЯ продукции к ним не относятся (подробнее об этом говорится ниже). Автор включил обсуждение этих процессов в статью лишь с точки зрения дополни-тельной иллюстрации ошибок в отнесении некоторых из процессов к специальным как таковым.
8 Внекоторых стандартах на системы менеджмента качества об-суждаемое положение из разряда рекомендации переведено в катего-рию ПРЯМОГОтребования. Например, в п. 7.5.2.1 стандарта ISO/TS 16949:2009 указано: требования раздела 7.5.2 должны применяться ко всем процессам производства и сервисного обслуживания
1. Валидация ISO
2. Чем отличается валидация от верификации?
3. Валидация документов
4. Валидация XML и XHTML
5. GMP валидация
6. Что такое валидация ИПДО?
Валидация - это придание законной силы, утверждение, легализация, ратификация (общегражданское право);
Валидация - это , позволяющий определить, насколько точно с позиций потенциального пользователя некоторая модель представляет заданные сущности реального мира (системное программирование);
Валидация - это процедура, дающая высокую степень уверенности в том, что конкретный процесс , метод или система будет последовательно приводить к результатам, отвечающим заранее установленным критериям приемлемости; в частности, валидация технологических процессов проводится с использованием образцов не менее трех серий реального товара с целью доказательство и предоставление документального свидетельства, что процесс (в пределах установленных параметров) обладает повторяемостью и приводит к ожидаемым результатам при производстве полупродукта или готового товара требуемого качества; валидация аналитических методов состоит в определении: точности, воспроизводимости, чувствительности, устойчивости (межлабораторная воспроизводимость), линейности и других метрологических характеристик
Валидация ISO
Применительно к системам менеджмента качества согласно стандартам ISO серии 9000:
Валидация - подтверждение на основе представления объективных свидетельств того, что требования, предназначенные для конкретного использования или применения, выполнены (ISO 9000:2005)
Валидация - подтвержение путем экспертизы и представления объективного доказательства того, что особые требования, предназначенные для конкретного применения, соблюдены.
Примечания:
1. При проектировании и разработке утверждение означает проведение экспертизы продукции с целью определения соответствия нуждам приобретателя.
2. Утверждение обычно осуществляется на конечной продукции в определенных условиях эксплуатации. Оно может быть необходимо на более ранних стадиях.
3. Термин «утверждено» используется для обозначения соответствующего статуса.
4. Могут осуществляться многократные утверждения, если предполагается различное использование. (ISO 8402:1994, п.2.18)
Анализ требований стандарта ISO 9001:
ISO 9001, п. 7.3.6: валидация проекта и разработки должна осуществляться в соответствии с запланированными мероприятиями, чтобы удостовериться, что полученная в результате продукция соответствует требованиям к установленному или предполагаемому использованию.
ISO 9001, п. 7.5.2: валидация процессов производства и обслуживания. должна подтверждать все процессы производства и обслуживания, результаты которых нельзя проверить посредством последовательного мониторинга или измерения. К ним относятся все процессы, недостатки которых становятся очевидными только после начала использования продукции или после предоставления услуги. Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов.
ISO 9000, примечание 3 п. 3.4.1: , в котором подтверждение соответствия конечной продукции затруднено или экономически нецелесообразно, часто относят к "специальному процессу".
Общепринятые требования к специальным производственным процессам, обеспечивающие их валидацию:
1) аттестация производственного процесса (технология, методика, рабочие инструкции...)
2) аттестация производственного оборудования (калибровка сварочных машин или роботов, краскопультов и систем подачи краски...)
3) аттестация материалов (электроды, газ, флюсы, краска, растворители, грунты...)
4) аттестация персонала (квалификационные требования к сварщикам или операторам сварочных роботов, наладчикам, сервисным компаниям...)
с соответствующим документальным подтверждением.
Спец. (СП) должен быть в управляемых условиях.
Управляемые условия включают:
Наличие информации, описывающей характеристики продукции и СП;
Наличие нормативной, конструкторской и технологической документации;
Использование пригодного оборудования;
Наличие и использование средств контроля и измерений;
Проведение контроля, измерений и испытаний;
Осуществление деятельности по выполнению СП;
Наличие квалифицированного и аттестованного персонала осуществляющего СП;
Повторную валидацию;
Наличие записей, содержащих достигнутые результаты или свидетельства осуществленной деятельности при выполнении СП.
Чем отличается валидация от верификации?
Верификация - подтверждение на основе представления объективных свидетельств того, что установленные требования были выполнены.
Валидация - подтверждение на основе представления объективных свидетельств того, что требования, предназначенные для конкретного использования или применения, выполнены.

Уже перевод с английского этих терминов дает определенную пищу для понимания разницы: verification - проверка, validation - придание законной силы.
Чтобы было проще понять, сразу приведу пример типичной верификации: тестирование программы или проведение испытания оборудования. Имея определенные требования на руках, мы проводим испытание товара и фиксируем, соблюдены ли требования. Результат верификации - это ответ на вопрос "Соответствует ли требованиям?".
Но далеко не всегда товар, соответствующий установленным требованиям, можно применять в конкретной ситуации. Например, лекарство прошло все положенные испытания и поступило в продажу. Значит ли это что оно может быть применено каким-то конкретным больным? Нет, т.к. каждый пациент имеет свои особенности и конкретно для этого лекарство может быть губительным, т.е. кто-то (врач) должен подтвердить: да, этому больному можно принимать это лекарство. То есть врач должен выполнить валидацию: придать законную силу конкретному применению.
Или еще пример. выпускает трубы, предназначенные для закладки в землю, в соответствии с некоторыми ТУ (Техническими условиями). Продукция этим ТУ соответствует, но поступил заказ, предполагающий укладку труб по дну моря. Могут ли трубы, соответствующие имеющимся ТУ, быть применены в данном случае? Именно валидация и дает ответ на этот вопрос.

Нетрудно видеть, что еще одно отличие состоит в том, что верификация производится всегда, а вот необходимость в валидации может и отсутствовать. Она появляется только тогда, когда возникают требования, связанные с конкретным применением продукции. Если фармацевтический завод выпускает лекарства, то он будет проверять лишь их соответствие требованиям, а проблемами применения конкретных лекарств конкретными пациентами заниматься не будет. Или тот же АвтоВАЗ.
Таким образом, можно констатировать следующее:
верификация - проводится практически всегда, выполняется методом проверки (сличения) характеристик продукции с заданными требованиями, результатом является вывод о соответствии (или несоответствии) продукции,
Валидация - проводится при необходимости, выполняется методом анализа заданных условий применения и оценки соответствия характеристик продукции этим требованиям, результатом является вывод о возможности применения продукции для конкретных условий.
Стандарт ИСО 9001 в двух местах обращается к этим терминам. Проверим, соответствует ли данное мной толкование содержанию разделов 7.3.5, 7.3.6 и 7.5.2.
"7.3.5. Верификация проекта и разработки. Верификация должна осуществляться в соответствии с запланированными мероприятиями (п. 7.3.1), чтобы удостовериться, что выходные данные проектирования и разработки соответствуют входным требованиям:".
"7.3.6. Валидация проекта и разработки. Валидация проекта и разработки должна осуществляться в соответствии с запланированными мероприятиями (п. 7.3.1), чтобы удостовериться, что полученная в результате продукция соответствует требованиям к установленному или предполагаемому использованию, если оно известно. Где это практически целесообразно, валидация должна быть завершена до поставки или применения продукции".
Нетрудно видеть, что трактовка находится в полном согласии с текстом этих разделов. При этом хотелось бы обратить внимание на то, что в п. 7.3.5 говорится о соответствии выходных данных , а в п. 7.3.6 - продукции. Это существенно! Это означает, что валидация проводится не для выходных данных , а для разработанной под конкретные условия продукции. Скажем, в деятельности института по разработке типовых проектов жилых зданий валидация не требуется - только верификация. А вот для деятельности по разработке проекта строительства жилого здания по тому же типовому проекту, но в конкретном месте, валидация уже необходима.
"7.5.2. Валидация процессов производства и обслуживания. Компания должна подтверждать все процессы производства и обслуживания, результаты которых нельзя проверить посредством последовательного мониторинга или измерения. К ним относятся все процессы, недостатки которых становятся очевидными только после начала использования продукции или после предоставления услуги. Валидация должна продемонстрировать способность этих процессов достигать запланированных результатов".

В технике или в системе менеджмента качества валидация подтверждает, что требования внешнего приобретателя или пользователя товара, услуги или системы удовлетворены. Верификация — это обычно внутренний процесс управления качеством, обеспечивающий согласие с правилами, стандартами или спецификацией. Простой способ запомнить разницу между валидацией и верификацией заключается в том, что валидация подтверждает, что «вы создали правильный товар», а верификация подтверждает, что «вы создали товар так, как и намеревались это сделать».
Валидация документов
Валидным является такой веб-документ, который прошел подобную процедуру и не имеет замечаний по коду. Код веб-страницы должен подчиняться определенным правилам, которые называются спецификацией, ее разрабатывает W3 (www.w3c.org) при поддержке разработчиков браузеров.
На первый взгляд, кажется, что валидация необходима, ведь речь идет о сокращении количества ляпов разработчиков и написании «правильного» кода. На деле все обстоит гораздо сложнее и вокруг валидации до сих пор ведутся горячие споры об ее актуальности. Чтобы объективно раскрыть этот вопрос далее рассмотрим плюсы и минусы такой проверки.
Хотя HTML-код имеет достаточно простую иерархическую структуру, при разрастании объема документа в коде легко запутаться, следовательно, просто и совершить ошибку. Браузеры, несмотря на явно неверный код, в любом случае постараются отобразить веб-страницу. Но поскольку единого регламента не существует о том, как же должен быть показан «кривой» документ, каждый браузер пытается сделать это по-своему. А это в свою очередь приводит к тому, что один и тот же документ может выглядеть по-разному в популярных браузерах. Исправление явных промахов и систематизация кода приводит, как правило, к стабильному результату.
Времена, когда производители браузеров добавляли уникальные возможности в свой товар вопреки всем стандартам, начинают уходить в прошлое. Каждая новая версия браузера все больше поддерживает спецификации и отображает документы с минимальными ошибками или вообще без них. Разработчики сайтов, также придерживающихся канонов веб-стандартов, таким образом соответствуют современным тенденциям развития веб-технологий.

Не стоит забывать и об XML (eXtensible Markup Language, расширяемый язык разметки). Этот язык становится стандартом де-факто для хранения данных и обмена информацией между разными приложениями. Синтаксис XML более жесткий, чем HTML и не прощает малейших ошибок. В каком-то смысле XML похож на языки программирования, в которых программа не будет скомпилирована, пока код не отлажен. HTML является первой ступенькой к изучению XML, поэтому приучая себя писать код по всем правилам, будет легче перейти к следующему этапу развития HTML.
Как это не удивительно, но среди веб-разработчиков тоже существует своя мода. Текущая мода — создавать валидные документы и вывешивать специальный значок в виде картинки, что сайт соответствует спецификации HTML. Подобная тенденция затронула даже заказчиков сайтов и при написании технического задания на разработку сайта некоторые из них специально оговаривают, чтобы сайт был выполнен по веб-стандартам.
Следование стандартам во многом дает множество выгод, которые проявляются в мелочах и становятся заметными при достижении определенной критической массы. В частности, объем кода становится меньше, компактнее и читабельнее. Соответственно, для пользователей повышается скорость загрузки сайта в целом.
Сайты, конечно же, делают для того, чтобы их посещали люди. Именно посетители выступают мерилом работы сайта, а их интересует и способ ее получения. Пользователь желает, чтобы сайт корректно отображался в его любимом браузере, быстро загружался и содержал те материалы, которые ему нужны. Заметьте, в этом списке нет ничего про код документа и его валидность, посетителей это просто не интересует. Поэтому совершенно невалидный сайт, но выполненный с душой, наполненный интересными материалами привлечет к себе больше посетителей, чем пустой ресурс, но сделанный по всем «правилам».
Разработчики браузеров не всегда следуют спецификации и в некоторых случаях трактуют код не по заданным правилам, а по-своему. В конечном итоге это приводит к тому, что веб-страница, которая правильно (т.е. так, как и задумывали разработчики) отображается в одном браузере, выводится с ошибками в другом. Следование спецификации в подобных случаях, скорее всего, отпугнет пользователей некоторых браузеров. К примеру, Internet Explorer (IE) в настоящее время занимает лидирующее положение среди браузеров, но при этом поддерживает спецификацию HTML и CSS хуже, чем Firefox и Opera. Очевидно, что пользователи IE при посещении сайта выполненного по всем стандартам, но не учитывающего специфику этого браузера, увидят неприглядную картину.
Заказчикам сайта, а также их разработчикам подобная ситуация не по нраву, поэтому стоя перед выбором: стандарты или браузер, они в большинстве своем выбирают браузер.
Получается неутешительная картина — тратить время на отладку кода для соответствия спецификации нет особой нужды. Это время лучше посвятить тому, чтобы документ без проблем работал в разных браузерах — так в основном размышляют веб-разработчики.
Валидация XML и XHTML
Для того, чтобы произвести валидацию содержимого, его сначала нужно получить. Источник (то, что мы должны проверить на соответствие набору определённых правил) может быть совершенно непредсказуемым:
Удалённый источник.
Абстрагируемся от указанных выше источников, ведь на самом деле нам не так уж и важно, откуда мы получаем данные для валидации: все они в конечном итоге предстают в виде строки. После того, как мы получили строку, нам нужно её обработать таким образом, чтобы получить элементы, с которыми мы можем работать на том языке программирования, на котором мы программируем. Для начала мы должны определиться, в какие сущности мы можем превратить входные данные. Вспоминаем, что основной единицей в XML/XHTML является элемент. От него и будем отталкиваться. Помимо элемента, нам нужен контейнер элементов, который мы будем называть документом.

Каждый XML/XHTML-документ состоит из набора элементов, причём всегда есть корневой элемент, содержащий в себе все остальные элементы. Постойте! Но ведь это же обычное дерево! Да-да, всё правильно: мы видим перед собой дерево элементов. Мы пришли к достаточно важному выводу: любой XML-документ (и документ на любом XML-подобном языке) можно представить в виде дерева. После, с этим деревом мы можем выполнять самый разный набор операций: сравнение, удаление, перестановка, траверсинг (операция прохода по всем узлам дерева) и другие.
Проверка XML несравненно проще проверки XHTML: нам необходимо лишь удостовериться в выполнении нескольких требований, что можно сделать совершенно спокойно, учитывая то, что у нас есть дерево элементов. Систематизируем необходимые правила:
Первый элемент в документе — это всегда декларация заголовка XML вида, где […] — атрибуты заголовка XML;
Все элементы должны быть названы верным образом и не должны содержать посторонних символов (пробелов, к примеру);
Все атрибуты должны быть записаны в правильной форме (проверяется достаточно просто, тем же регулярным выражением);
Документ должен содержать только один корневой элемент;
Вложенность элементов должна быть соблюдена (проверка данного утверждения достигается за счёт использования стека элементов, с помощью которого мы проверяем соответствия открывающих и закрывающих элементов);
Если все вышеуказанные правила соблюдаются, то документ считается валидным XML-документом. В противном случае — документ содержит ошибки, список которых валидатор может вывести пользователю для ознакомления и исправления.
Проверка XHTML основывается на валидации XML. Сначала мы должны удостовериться в том, что документ является валидным с точки зрения XML (то есть соблюдена вложенность тегов, правильно оформлены элементы и их атрибуты, и другие), и уже потом накладывать дополнительные правила. Если документ не является валидным с точки зрения XML, то он заведомо не является валидным и с точки зрения XHTML.
Чтобы применять какие-либо правила XHTML к документу, сначала нужно эти правила описать таким образом, чтобы их было легко получить и как трафарет наложить на документ. Для валидации XHTML-документов правила могут храниться в виде нескольких форматов:
DTD-документы;
Независимо от формата, производится следующий набор проверок:
Проверка всех используемых элементов в документе на их наличие в XHTML (если элемент, указанный в документе, не существует, то выдаётся соответствующая ошибка);
Проверка на наличие обязательных атрибутов у соответствующих элементов;
Проверка типа содержимого некоторых атрибутов на соответствие тем типам, которые указаны в правилах;
Тип содержимого элемента должен совпдадать с тем, который указан в правилах;
Так как XHTML определяет классы элементов (блоковые и текстовые), то валидатор должен убедиться в том, что элементы одного класса (уровня) должны быть правильно вложены в элементы другого класса (уровня). Подобные закономерности также описываются в правилах.
После выполнения указанного набора правил можно говорить о том, является ли документ валидным или напротив: содержит какие-то ошибки, которые валидатор может указать пользователю.
GMP валидация
GMP является сложным комплексным решением проекта, строительства, эксплуатации и компании производства, требует большое финансовое и трудовое вложение. Это уже стало обязательно достигнутым уровнем для фармацевтического завода.
На практике реальные опыты и профисиональные консультации могут помочь предотвратить ошибки в проектировании и комплектации оборудования, которые могут вызывать огромные невозмещаемые потери в капиталовложении, времени и силы.
Для работающего завода: обследывание завода в отношении здания, общетехнического условия, ассортименте продукции и соответствующих цехов, состояния оборудования, структуры и персонала, и занния и навыки персонала о правилах GMP, документации о контроле качества и менеджменте, анализуя степень соответствия с требованием действующиего стандарта GMP, составят общий план работы усовершенствия и расписание. Две стороны подверждают и начинается осуществовать.

Для нового объекта: планировка завода: общий план, проект технологии, складирования, качества, процесса производства и дает замечание или советы. Проверяет выбор оборудования и поставщика
Акретитация по правилам GMP не только нуждается в ссответствующих соружении и оборудовании, но и системе документации: обязанности отделов и должностей, правила менеджмента , правила операций, правила контроля качества, стандарта качества, записи серий производства.
Что такое валидация ИПДО?
Валидация ИПДО является механизмом, нацеленным на обеспечение качества и является неотъемлемой частью процесса ИПДО. Она несет две основные функции. Во-первых, она стимулирует диалог и процесс обучения на уровне страны. Во-вторых, она поддерживает уровень единого глобального стандарта ИПДО во всех внедряющих странах. Валидация не является аудитом. Она не является повторением процесса раскрытия и сравнения, которые составляют часть процесса подготовки отчетов ИПДО. У валидации более всеобъемлющие цели: при содействии заинтересованных сторон она оценивает внедрение ИПДО; оценивает результаты на соответствие глобальному стандарту; и изыскивает возможности укрепления развития процесса ИПДО.
Кроме того, валидация является механизмом, который применяется для определения статуса страны как страны-кандидата или страны , удовлетворяющей ИПДО. В настоящий момент 23 страны являются кандидатами. Все эти страны удовлетворяют четырем вступительным требованиям и находятся на различных стадиях внедрения ИПДО. ИПДО требует для оценки соответствия ИПДО, чтоб страны завершили валидационный процесс в двухлетний срок.
Посредством валидации страны, демонстрирующие свое соответствие с требованиями ИПДО (или заметный прогресс в достижении этой цели) получают международное признание своих усилий и достижений. Если валидация не завершена или она показывает отсутствие заметного прогресса в достижении соответствия требованиям ИПДО, Правление ИПДО отзывает статус страны-кандидата.
Процесс валидации проходит на международном уровне и контролируется мультистейкхолдерной группой на уровне государства. валидации изложена в Правила ИПДО, в том числе руководство по валидации.
Первым шагом является назначение мультистейкхолдерной группой валидатора. Правление ИПДО составило список аккредитованных валидаторов ИПДО, и подготовило указания для внедряющих стран о назначении валидатора.

Выбранный валидатор пользуется тремя основными документами в своей работе.
Таковыми являются:
Рабочий план страны
Валидационные требования и Метод оценки показателей, и
Анкеты компаний
Пользуясь данными документами валидатор встречается с мульти-стейкхолдерной группой, привлекается компанией для сверки данных раскрытых компаниями, правительством и другими ключевыми стейкхолдерами (включая организации и общественность, не вошедшие в состав мульти-стейкхолдерной группы).
Пользуясь данными сведениями валидатор завершает отчет, включающий:
Сжатый обзорный отчет о прогрессе в выполнении рабочего плана страны;
Сжатый обзорный отчет о прогрессе в достижении показателей валидационного графика;
Заполненный валидационный график;
Обзорный отчет о внедрении компаниями;
Совокупные анкеты компаний;
Общая оценка внедрения ИПДО: является ли страна кандидатом, соответствует требованиям, есть или нет заметного прогресса.

Данный отчет сначала поступает в адрес мультистейкхолдерной группы, правительства и Правления ИПДО. Если данные группы согласовывают валидационный отчет, он публикуется и замечания принимаются к исполнению. Если возникает разногласия относительно валидационного процесса, тогда он рассматривается сначала на местном уровне. Правление ИПДО привлекается только в случае возникновения серьезных разногласий.
Источники
ru.wikipedia.org Википедия - вободная энциклопедия
htmlbook.ru Для тех, кто делает сайты
certicom.kiev.ua СертиКом
enumerate.ru В поисках упорядоченной истины
luxunig.com Фармацевтический инженеринг
Энциклопедия инвестора . 2013 .
Синонимы :Смотреть что такое "Валидация" в других словарях:
Валидация - придание законной силы, утверждение, легализация, ратификация. Словарь бизнес терминов. Академик.ру. 2001 … Словарь бизнес-терминов
Понятия, которые мы будем основательно разбирать, довольно часто встречаются как в обыденной жизни, так и в специализированной литературе, профессиональной деятельности. Многие хотят знать, верификация и валидация - что это простыми словами? В чем разница между этими терминами? Давайте порассуждаем вместе.
Валидация и верификация - что это простыми словами?
Оба понятия связаны с тестированием какого-либо продукта и обеспечением его качества. Если мы будем говорить простым языком, то выведем следующее:
- Валидация - гарантированная уверенность производителя в том, что он создал продукт по всем необходимым стандартам.
- Верификация - помогает увериться в том, что изделие соответствует всем изначально заданным требованиям к нему.
Рассказывая простыми словами, что это - верификация и валидация, нужно сделать упор и на такие факты:
- Для потребителя важнее всего валидация - уверенность в том, что он получает правильный продукт, соответствующий его требованиям.
- Для производителя более ценной будет верификация - подтверждение того, что изделие, которое он отправляет на реализацию, отвечает всем необходимым стандартам и нормам.
Еще одно значение
Мы еще разберем различие в понятиях "верификация" и "валидация" в тестировании. Ведь по большому счету они связаны с международными требованиями к проверке, приемке технологий и различной продукции.
Однако вместе с тем слова плотно вошли в жизнь и интернет-пользователей. Например, регистрируясь в платежных системах типа "Киви", "Яндекс. Деньги", вы должны пройти процесс верификации. В данном случае это обозначает проверку подлинности указанных данных о себе, идентификацию вас системой.

А те, кто активно пользуются социальными сетями ("ВКонтакте", "Одноклассники" и проч.), рано или поздно видят перед собой окошко с просьбой пройти валидацию. Это такая же проверка истинности введенных вами данных. К примеру, на привязанный к аккаунту телефон приходит СМС с кодом, который нужно напечатать в определенное поле, чтобы подтвердить, что вы являетесь владельцем указанного номера.
Таким образом, в данном случае трудно выделить разницу между валидацией и верификации. И то и другое, по сути, здесь является проверкой на указание соответствующих действительности данных. Хотим также указать на факт, что валидацию/верификацию успешно используют разработчики различных вирусов с целью выманивания у вас личной информации. Отчего такие данные следует вводить на надежных ресурсах, с компьютера, защищенного современным качественным антивирусом.
Определение стандарта ИСО 9000:2000
Объяснить простыми словами, что это - верификация и валидация, поможет характеристика этих терминов, данная в документах ИСО (ISO - Международная организация по стандартизации). Здесь мы видим следующее:
- Верификация - подтверждение на основе объективных предоставленных фактов того, что установленные нормы были выполнены.
- Валидация - подтверждение на основе объективных предоставленных фактов того, что установленные нормы для конкретного применения выполнены.

Вот из этих определений уже вытекает разница валидации и верификации:
- Первая процедура проводится только по необходимости. Продукт анализируется в заданных условиях эксплуатации. Результатом будет вердикт: возможно ли его использовать в данной обстановке.
- Вторая процедура практически обязательна. Это проверка на соответствие продукта требованиям, которые будут актуальны при любых условиях, при любом использовании.
Прочие определения верификации
Помочь разобраться в теме нам поможет ряд распространенных определений рассматриваемых понятий. Приведем характеристики верификации:
- Подтверждение соответствия выпущенного товара, продукта определенным эталонам.
- Практически обязательная процедура; сличение характеристик произведенной единицы с рядом заданных требований. Результат - вердикт соответствия или несоответствия последним.
- Провозглашение подтверждения, что установленные нормы в отношении изделия были выполнены.
- Простыми словами - создан продукт, который соответствует необходимым стандартам.

Прочие определения валидации
Рассмотрим теперь определения валидации:
- Практическое определение того, насколько тот или иной продукт соответствует ожиданиям его непосредственных пользователей.
- Процедура, которую проводят при необходимости. Это распространенный анализ заданных условий и оценка характеристик продукта касательно его эксплуатации в данной среде. Результат - вывод о возможности использования товара, изобретения в определенной сфере.
- Подтверждение соблюдения требований системы стандартов, заказчика, непосредственного пользователя и проч.
- Простыми словами - создан правильный продукт, удовлетворяющий потребителя.
Отличия на основе перевода
Определить, в чем разница между валидацией и верификацией, поможет и обращение к переводу этих слов, имеющих английские корни:

- Verification - какая-либо проверка.
- Validation - придание чему-либо законной силы.
Даже из этого следует, что верификация предшествует валидации, не является конечной. Окончательный вердикт продукту, имеющий законную силу, дает именно последняя.
Отличия верификации и валидации в сравнении
В сравнительной таблице легче обозначить различия этих в чем-то схожих терминов.
| Верификация | Валидация |
| Делаем ли мы продукцию правильно? | Произвели ли мы правильный продукт? |
| Вся ли функциональность была реализована? | Верно ли функциональность была реализована? |
| Верификация предшествует валидации: она включает в себя полную проверку правильности написания, производства и прочего сотворения. | Случается уже после верификации - качества произведенного продукта. |
| Проводят разработчики. | Проводят тестировщики. |
| Статистический тип анализа: сравнение с установленными требованиями к продукту. | Динамический тип анализа: продукт тестируется в эксплуатации для выяснения его соответствия нормам. |
| Объективная оценка: выносится на основе соответствия определенным стандартам. | Субъективная оценка: личная оценка, которую ставит специалист-тестировщик. |
Давайте еще немного порассуждаем, чем отличается валидация от верификации, в следующем разделе.
Ключевые различия понятий
Итак, расставим все точки над i. Верификация - это любое тестирование, через которое проходит продукт. Проверка правильности технологии его производства, а также качества изделия. Валидация же - понятие, более близкое к аттестации. Это соответствие каким-то конкретным, а не общим требованиям. Насколько хорош продукт не вообще, а именно для определенного потребителя, заказчика или заданных условий.
Еще можно отметить, что верификация - это бумажное, теоретические тестирование технологии или продукта. Валидация же - реальная, физическая проверка, осуществляемая на практике, в конкретных условиях.
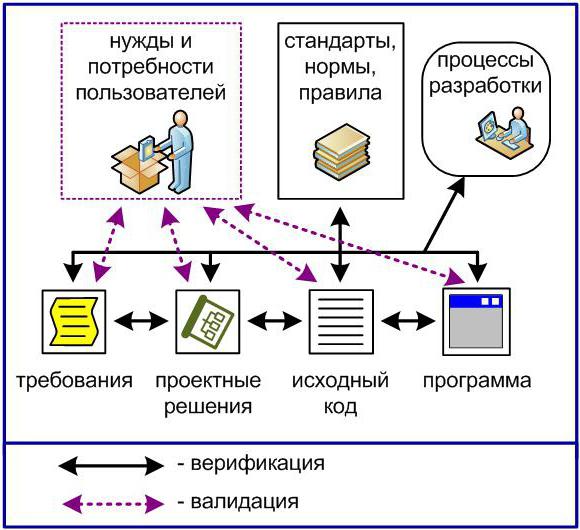
Если изделие прошло верификацию, значит, оно соответствует каким-то заданным технологическим требованиям. Если же успешно пройдена валидация, выходит, что на практике оно также без нареканий применимо. Отсюда можно вынести, что последнее понятие несколько важнее, показательнее, нежели первое.
Примеры верификации
Давайте посмотрим на конкретные примеры, чтобы закрепить в голове разницу между этими понятиями.
Фармацевтический завод проверяет лекарства на соответствие конкретным требованиям. На вводе в производство устанавливается их безопасность для пациента в определенных дозах, отсутствие эффекта плацебо, неимение возможности проявления губительного привыкания и проч. Таким образом, верификация препаратом пройдена. А валидацию в этом случае проводит уже лечащий доктор: он определяет, поможет ли лекарство конкретному пациенту, не приведет ли его применение к риску для жизни и здоровья этого человека и т. д.
Рассмотрим на примере велосипеда. Проверяем, есть ли руль, сидение, цепи, колеса, тормозная система и проч. Все на месте? Верификация пройдена!
Примеры валидации
Теперь примеры, чем отличается валидация от верификации.

Какое-либо предприятие в соответствии с определенными требованиями производит универсальные трубы. Поступает вопрос от заказчика: возможно ли данный продукт проложить по дну моря? Производитель должен провести валидацию своих труб в соответствии с предложенными условиями, чтобы объективно ответить на этот вопрос.
На примере того же велосипеда рассмотреть валидацию тоже очень легко. На устройстве можно кататься? Можно затормозить? Можно повернуть вправо, влево? Переключить скорость? Если все возможно, валидация пройдена. Не смогли затормозить, упало сидение, расшатан руль - увы, велосипед данную процедуру не прошел.
Вот мы и разобрали понятия "верификация" и "валидация", постаравшись выразить все простым языком. Надеемся, что это поможет вам четко проследить разницу между ними, особенности каждого.





